

Aimovig Inj Lös 140 мг/мл попередньо заповнена ручка 1 мл
Aimovig Inj Lös 140 mg/ml Fertigpen 1 ml
-
89408.43 RUB
Дата доставки:
25.12.2025 - 08.01.2026
При оплате криптовалютой:
Ваша прибыль 8940.84 RUB / 105.53 USDT

- Наявність: В наявності
- Виробник: NOVARTIS SCHWEIZ AG
- Модель: 7747988
- ATC-код N02CD01
- EAN 7680667480037
Склад:
Наведіть телефон на qr-код

Опис
 Deutsch
Deutsch French
French Italian
ItalianWas ist Aimovig und wann wird es angewendet?
Aimovig enthält den Wirkstoff Erenumab. Erenumab ist ein humaner monoklonaler Antikörper. Monoklonale Antikörper sind Eiweisse, die bestimmte Eiweisse im Körper erkennen und an sie binden. Aimovig gehört zu einer Gruppe von Arzneimitteln, die als Antikörper gegen den CGRP (Calcitonin Gene-Related Peptide) Rezeptor eingesetzt werden.
Aimovig wirkt, indem es die Aktivität des CGRP-Moleküls, welches mit Migräne in Zusammenhang gebracht worden ist, blockiert. Aimovig wird zur Behandlung der Migräne bei Erwachsenen verwendet, bei denen eine vorbeugende Therapie angezeigt ist. Aimovig wird auf Verschreibung des Arztes oder der Ärztin angewendet.
Wann darf Aimovig nicht eingenommen / angewendet werden?
Aimovig darf nicht angewendet werden, wenn Sie allergisch gegen Erenumab oder einen der in Aimovig enthaltenen Hilfsstoffe (aufgeführt unter «Was ist in Aimovig enthalten») sind.
Die Anwendung von Aimovig wurde bei Kindern und Jugendlichen unter 18 Jahren nicht untersucht. Es ist nicht bekannt, ob Aimovig bei Kindern und Jugendlichen sicher und wirksam ist. Aimovig darf daher bei dieser Altersgruppe nicht angewendet werden.
Wann ist bei der Einnahme / Anwendung von Aimovig Vorsicht geboten?
Wenn bei Ihnen Symptome einer schwerwiegenden allergischen Reaktion, wie z.B. Ausschlag (schweres Jucken der Haut mit rotem Ausschlag oder Erhebungen, sogenannte Nesselsucht) oder Schwellungen, die in der Regel Gesicht, Mund, Zunge oder Rachen betreffen, oder Atembeschwerden auftreten beenden Sie sofort die Anwendung von Aimovig und informieren Sie sofort Ihren Arzt bzw. Ihre Ärztin oder rufen Sie den Notarzt. Schwerwiegende allergische Reaktionen können innerhalb von Minuten auftreten, einige können jedoch auch erst mehr als eine Woche nach der Anwendung von Aimovig in Erscheinung treten.
Unter Aimovig kann es zu Verstopfung mit schweren Komplikationen kommen. Teilen Sie Ihrem Arzt/Ihrer Ärztin mit, wenn Sie an Verstopfung leiden, in der Vorgeschichte an Verstopfung gelitten haben oder gleichzeitig Medikamente einnehmen, welche zu Verstopfung führen können. Ihr Arzt/Ihre Ärztin wird Sie bei Anzeichen schwerwiegender Verstopfung überwachen und entsprechend behandeln.
Die abnehmbare Kappe des Aimovig-Fertigpens enthält trockenen Naturkautschuklatex, der bei Personen mit Latexempfindlichkeit allergische Reaktionen hervorrufen kann. Informieren Sie Ihren Arzt bzw. Ihre Ärztin, wenn Sie auf Latex allergisch sind.
Bisher liegen keine Hinweise vor, dass die Verkehrstüchtigkeit und das Bedienen von Maschinen durch die Anwendung von Aimovig beeinträchtigt wird.
Dieses Arzneimittel (Fertigpen à 70 mg und 140 mg) enthält weniger als 1 mmol Natrium (23 mg) pro Fertigpen d.h. es ist nahezu «natriumfrei».
Informieren Sie Ihren Arzt, Apotheker bzw. Ihre Ärztin, Apothekerin, wenn Sie
- an anderen Krankheiten leiden,
- Allergien haben oder
- andere Arzneimittel (auch selbst gekaufte!) einnehmen.
Darf Aimovig während einer Schwangerschaft oder in der Stillzeit eingenommen / angewendet werden?
Aimovig wurde bei Schwangeren nicht untersucht. Es ist nicht bekannt, ob Aimovig dem ungeborenen Kind schadet, und deshalb sollte Aimovig während der Schwangerschaft nicht angewendet werden. Informieren Sie Ihren Arzt bzw. Ihre Ärztin, wenn Sie versuchen, schwanger zu werden, oder denken, möglicherweise schwanger zu sein, während Sie Aimovig anwenden. Es ist nicht bekannt, ob Aimovig in die Muttermilch übergeht. Da viele Arzneimittel, darunter Antikörper, in die Muttermilch ausgeschieden werden, kann ein Risiko für das Neugeborene/Kleinkind nicht ausgeschlossen werden. Es ist wichtig, Ihren Arzt bzw. Ihre Ärztin zu informieren, wenn Sie stillen oder vorhaben zu stillen. Ihr Arzt bzw. Ihre Ärztin wird entscheiden, ob Sie das Stillen oder eher die Anwendung von Aimovig beenden sollten.
Wie verwenden Sie Aimovig?
Die Indikation für die Therapie muss durch einen Arzt oder eine Ärztin mit Erfahrung auf dem Gebiet der Migränebehandlung gestellt werden. Sie werden durch diesen Arzt oder diese Ärztin in der weiteren Behandlung begleitet werden.
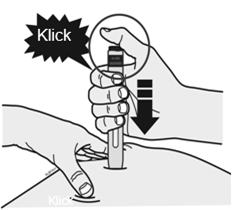
Aimovig wird als Injektion unter die Haut («subkutan») verabreicht.
Sie und Ihr Arzt bzw. Ihre Ärztin entscheiden, ob Sie sich Aimovig selbst injizieren sollen.
Es ist wichtig, dass Sie nicht versuchen, die Injektion selbst durchzuführen, ohne vorher ein Training durch Ihren Arzt oder Apotheker bzw. durch Ihre Ärztin oder Apothekerin oder durch eine medizinische Fachperson erhalten zu haben.
Hinweise zur richtigen Vorbereitung und Verabreichung Ihrer Aimovig-Injektionen zuhause finden Sie in der ausführlichen Gebrauchsanweisung am Ende dieser Packungsbeilage.
Dosierung von Aimovig:
Die Dosis von Aimovig beträgt 70 mg einmal monatlich.
Bei Patienten, die auf diese Dosierung nach einigen Monaten eine ungenügende Wirkung zeigen, kann Ihr Arzt / Ihre Ärztin die Dosierung auf 140 mg einmal monatlich steigern, solange dadurch eine bessere Wirkung nachweisbar ist.
Ändern Sie nicht von sich aus die verschriebene Dosierung. Wenn Sie glauben, das Arzneimittel wirke zu schwach oder zu stark, so sprechen Sie mit Ihrem Arzt oder Apotheker bzw. mit Ihrer Ärztin oder Apothekerin.
Wie lange wird Aimovig angewendet?
Wenden Sie Aimovig so lange einmal im Monat an, wie Ihr Arzt bzw. Ihre Ärztin es anordnet.
Wenn Sie Fragen zur Anwendungsdauer von Aimovig haben, fragen Sie Ihren Arzt bzw. Ihre Ärztin.
Wenn Sie eine grössere Menge von Aimovig angewendet haben, als Sie sollten
Falls Sie eine höhere als die empfohlene Dosis von Aimovig angewendet haben, wenden Sie sich an Ihren Arzt bzw. Ihre Ärztin.
Wenn Sie die Anwendung von Aimovig vergessen haben
Wenn Sie die Anwendung von Aimovig vergessen haben, holen Sie dies so bald wie möglich nach. Setzen Sie sich dann mit Ihrem Arzt bzw. Ihrer Ärztin in Verbindung, der bzw. die Ihnen mitteilen wird, wann Sie Ihre nächsten Dosen verabreichen sollen. Halten Sie sich genau an den Zeitplan, den Ihr Arzt bzw. Ihre Ärztin Ihnen vorgegeben hat.
Wenn Sie die Anwendung von Aimovig abbrechen
Beenden Sie die Anwendung von Aimovig nicht, ohne zuerst den Rat Ihres Arztes bzw. Ihrer Ärztin einzuholen. Wenn Sie die Behandlung beenden, könnten Ihre Symptome erneut auftreten.
Wenden Sie sich an Ihren Arzt bzw. Ihre Ärztin, wenn Sie weitere Fragen zur Anwendung von Aimovig haben.
Kinder und Jugendliche (unter 18 Jahren)
Die Anwendung von Aimovig wurde bei Kindern und Jugendlichen unter 18 Jahren bisher nicht untersucht. Es ist nicht bekannt, ob Aimovig bei Kindern und Jugendlichen sicher und wirksam ist. Aimovig darf daher bei dieser Altersgruppe nicht angewendet werden.
Welche Nebenwirkungen kann Aimovig haben?
Es können schwerwiegende allergische Reaktionen wie z.B. Ausschlag (schweres Jucken der Haut mit rotem Ausschlag oder Erhebungen, sogenannte Nesselsucht) oder Schwellungen, die in der Regel Gesicht, Mund, Zunge oder Rachen betreffen, oder Atembeschwerden auftreten. Beenden Sie sofort die Anwendung von Aimovig und informieren Sie sofort Ihren Arzt bzw. Ihre Ärztin oder rufen Sie den Notarzt, wenn Sie solche Anzeichen bemerken. Ihr Arzt/Ihre Ärztin wird eine geeignete Therapie einleiten.
Es wurde über Fälle von Verstopfung mit schweren Komplikationen berichtet. Viele dieser Fälle wurden bei Patienten gemeldet, die Verstopfung in der Vorgeschichte hatten oder gleichzeitig Arzneimittel anwendeten, die zu Verstopfung führen können. Bei einigen schweren Fällen war ein Spitalaufenthalt erforderlich. Bitte informieren Sie daher Ihren Arzt, wenn Sie an Verstopfung leiden.
Weiter können die im Folgenden aufgeführten Nebenwirkungen auftreten. Wenn sich diese Nebenwirkungen verschlimmern, informieren Sie bitte Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin.
Häufig (betrifft 1 bis 10 von 100 Anwendern)
Anzeichen und Symptome von Schmerzen, Rötung, Schwellung, Juckreiz an der Injektionsstelle (Reaktionen an der Injektionsstelle)
- Verstopfung
- Muskelkrämpfe
- Juckreiz
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt, Apotheker oder Ihre Ärztin, Apothekerin. Dies gilt insbesondere auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind.
Was ist ferner zu beachten?
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Lagerungshinweis
Im Kühlschrank (2-8 °C) lagern.
Den Fertigpen in der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Nachdem Aimovig aus dem Kühlschrank genommen worden ist, kann es bei Raumtemperatur (15-25 °C) aufbewahrt werden und muss dann aber innerhalb von 14 Tagen angewendet werden. Aimovig darf nicht wieder im Kühlschrank aufbewahrt werden, nachdem es Raumtemperatur angenommen hat.
Verwenden Sie dieses Arzneimittel nicht, wenn die Flüssigkeit gut erkennbare Feststoffteilchen enthält, trüb oder eindeutig gelb ist.
Fragen Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, wie das Arzneimittel zu entsorgen ist, wenn Sie es nicht mehr verwenden.
Weitere Auskünfte erteilt Ihnen Ihr Arzt, Apotheker bzw. Ihre Ärztin, Apothekerin. Diese Personen verfügen über die ausführliche Fachinformation.
Was ist in Aimovig enthalten?
Wirkstoffe
Jeder Fertigpen zu 1 ml enthält 70 mg oder 140 mg Erenumab.
Hilfsstoffe
Saccharose, Polysorbat 80, Natriumhydroxid, Essigsäure 99%, Wasser für Injektionszwecke.
Zulassungsnummer
66748 (Swissmedic)
Wo erhalten Sie Aimovig? Welche Packungen sind erhältlich?
In Apotheken nur gegen ärztliche Verschreibung.
Packungen zu 1 Fertigpen à 70 mg oder 140 mg
Zulassungsinhaberin
Novartis Pharma Schweiz AG, Risch; Domizil: 6343 Rotkreuz
Diese Packungsbeilage wurde im August 2020 letztmals durch die Arzneimittelbehörde (Swissmedic) geprüft.
Gebrauchsanweisung für Aimovig 70 mg/ml und 140 mg/ml Injektionslösung in einem Fertigpen
Lesen Sie vor der Anwendung eines Aimovig-Fertigpens diese Anweisung vollständig durch. Es ist wichtig, dass Sie nicht versuchen, die Injektion selbst durchzuführen, ohne vorher ein Training durch Ihren Arzt oder Apotheker bzw. durch Ihre Ärztin oder Apothekerin oder durch eine medizinische Fachperson erhalten zu haben.
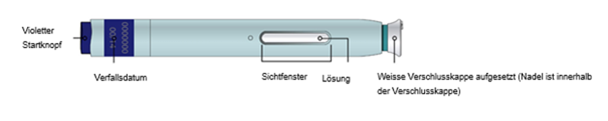
Darstellung des Aimovig 70-mg-Pen (mit hellblauem Körper, violettem Startknopf, weisser Verschlusskappe und grüner Sicherheitshülse)
Ansicht vor Anwendung |
|
Darstellung des Aimovig 140-mg-Pens (mit dunkelblauem Körper, grauem Startknopf, orangefarbener Verschlusskappe und gelber Sicherheitshülse) |
Ansicht vor Anwendung |
|
Wichtig: Nadel befindet sich innerhalb der grünen (70 mg Pen) oder gelben (140 mg Pen) Sicherheitshülse: |

| |
|
|
|
|
|
|
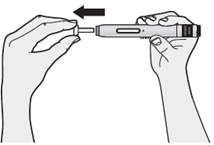
In all diesen Fällen müssen Sie einen neuen Fertigpen verwenden. Wenn Sie sich nicht sicher sind, wenden Sie sich an die für Sie zuständige medizinische Fachperson. | |
|
|
| |
|
|
|
Flach auseinanderziehen Ziehen Sie die Haut flach auseinander, indem Sie Ihren Daumen und Ihre Finger in entgegengesetzte Richtungen ziehen. Es sollte eine ungefähr fünf Zentimeter breite feste Stelle entstehen. ODER Zusammendrücken Drücken Sie die Haut zwischen Daumen und Fingern fest zusammen. Es sollte eine ungefähr fünf Zentimeter breite Stelle entstehen. |
|
|
|
|
|
|
|
|
| |










Qu’est-ce que le Aimovig et quand doit-il être utilisé?
Aimovig contient le principe actif érénumab. L'érénumab est un anticorps monoclonal humain. Les anticorps monoclonaux sont des protéines qui reconnaissent certaines protéines dans le corps et se lient à elles. Aimovig appartient à un groupe de médicaments qui sont utilisés comme anticorps contre le récepteur du peptide relié au gène de la calcitonine (CGRP, calcitonin gene-related peptide).
Aimovig agit en bloquant l'activité de la molécule CGRP, qui a été mise en rapport avec la migraine. Aimovig est utilisé pour le traitement de la migraine chez les adultes pour lesquels un traitement préventif est indiqué. Aimovig est utilisé selon prescription du médecin.
Quand Aimovig ne doit-il pas être pris/utilisé?
Aimovig ne doit pas être pris si vous êtes allergique à l'érénumab ou à l'un des excipients contenus dans Aimovig (mentionnés dans «Que contient Aimovig?»).
L'utilisation d'Aimovig n'a pas été étudiée chez les enfants et les adolescents de moins de 18 ans. On ignore si Aimovig est sûr et efficace chez les enfants et les adolescents. Aimovig ne doit donc pas être utilisé dans ce groupe d'âge.
Quelles sont les précautions à observer lors de la prise/de l’utilisation d’Aimovig?
Si des symptômes d'une réaction allergique sévère, comme p.ex. une éruption (démangeaison grave de la peau avec une éruption rouge ou des protubérances, ce que l'on appelle de l'urticaire) ou des gonflements qui touchent en général le visage, la bouche, la langue ou la gorge, ou des troubles respiratoires surviennent, arrêtez immédiatement d'utiliser Aimovig et informez immédiatement votre médecin ou appelez le médecin d'urgence. Des réactions allergiques sévères peuvent survenir en l'espace de quelques minutes, certaines peuvent toutefois également apparaître plus d'une semaine après l'utilisation d'Aimovig.
Aimovig peut provoquer des constipations accompagnées de graves complications. Informez votre médecin si vous souffrez de constipation, si vous avez des antécédents de constipation, ou si vous prenez simultanément des médicaments susceptibles de provoquer des constipations. Votre médecin vous surveillera et vous traitera en conséquence en présence de signes de grave constipation.
Le capuchon amovible du stylo prérempli Aimovig contient du latex de caoutchouc naturel sec qui peut provoquer des réactions allergiques chez les personnes sensibles au latex. Veuillez informer votre médecin si vous êtes allergique au latex.
Rien n'indique à ce jour que l'aptitude à la conduite et la capacité à utiliser des machines puissent être affectées par l'utilisation d'Aimovig.
Ce médicament (stylo prérempli à 70 mg et 140 mg) contient moins de 1 mmol de sodium (23 mg) par stylo prérempli, c.-à-d. qu'il est pratiquement «sans sodium».
Veuillez informer votre médecin ou votre pharmacien si
- vous souffrez d'une autre maladie
- vous êtes allergique
- vous prenez déjà d'autres médicaments (même en automédication!).
Aimovig peut-il être pris/utilisé pendant la grossesse ou l’allaitement?
Aimovig n'a pas été étudié chez les femmes enceintes. On ignore si Aimovig nuit à la santé de l'enfant à naître, Aimovig ne doit donc pas être utilisé pendant la grossesse. Veuillez informer votre médecin si vous prévoyez une grossesse ou si vous pensez être éventuellement enceinte pendant l'utilisation d'Aimovig. On ignore si Aimovig passe dans le lait maternel. Étant donné que de nombreux médicaments, entre autres les anticorps, sont éliminés dans le lait maternel, un risque pour le nouveau-né ou pour l'enfant en bas âge ne peut pas être exclu. Il est important d'informer votre médecin si vous allaitez ou si souhaitez allaiter. Votre médecin décidera si vous devez arrêter d'allaiter ou si vous devez plutôt arrêter l'utilisation d'Aimovig.
Comment utiliser Aimovig?
L'indication pour le traitement doit être posée par un médecin expérimenté dans le domaine du traitement de la migraine. Vous serez accompagné(e) par ce médecin pour la suite du traitement.
Aimovig est administré sous forme d'injection sous la peau («sous-cutanée»).
Vous et votre médecin déciderez si vous pouvez vous injecter vous-même Aimovig.
Il est important que vous ne tentiez pas de procéder vous-même à l'injection sans avoir reçu auparavant une formation de votre médecin, de votre pharmacien ou d'un spécialiste médical.
Vous trouverez les indications concernant la préparation et l'administration correctes de vos injections d'Aimovig à domicile dans la notice d'utilisation détaillée à la fin de cette notice d'emballage.
Dosage d'Aimovig:
La dose d'Aimovig est de 70 mg une fois par mois.
Si ce dosage ne s'avère pas suffisamment efficace après quelques mois, votre médecin peut augmenter le dosage à 140 mg une fois par mois, tant qu'une meilleure efficacité peut ainsi être démontrée.
Ne changez pas de votre propre chef le dosage prescrit. Adressez-vous à votre médecin ou à votre pharmacien si vous estimez que l'efficacité du médicament est trop faible ou au contraire trop forte.
Combien de temps Aimovig doit-il être utilisé?
Utilisez Aimovig une fois par mois aussi longtemps que votre médecin vous l'a ordonné.
Demandez à votre médecin si vous avez des questions concernant la durée d'utilisation d'Aimovig.
Si vous avez utilisé plus d'Aimovig que vous auriez dû
Si vous avez utilisé une dose d'Aimovig plus élevée que la dose recommandée, consultez votre médecin.
Si vous avez oublié d'utiliser Aimovig
Si vous avez oublié d'utiliser Aimovig, prenez la dose aussi vite que possible. Contactez votre médecin qui vous dira quand vous devrez prendre les doses suivantes. Respectez exactement les horaires de prise du médicament que votre médecin vous a prescrits.
Si vous arrêtez d'utiliser d'Aimovig
N'arrêtez pas d'utiliser Aimovig sans demander auparavant conseil à votre médecin. Si vous arrêtez le traitement, vos symptômes pourraient réapparaître.
Consultez votre médecin si vous avez d'autres questions concernant l'utilisation d'Aimovig.
Enfants et adolescents (de moins de 18 ans)
L'utilisation d'Aimovig n'a pas été étudiée à ce jour chez les enfants et les adolescents de moins de 18 ans. On ignore si Aimovig est sûr et efficace chez les enfants et les adolescents. Aimovig ne doit donc pas être utilisé dans ce groupe d'âge.
Quels effets secondaires Aimovig peut-il provoquer?
Des réactions allergiques sévères, comme p.ex. une éruption (démangeaison grave de la peau avec une éruption rouge ou des protubérances, ce que l'on appelle de l'urticaire) ou des gonflements qui touchent en général le visage, la bouche, la langue ou la gorge, ou des troubles respiratoires peuvent survenir. Arrêtez immédiatement d'utiliser Aimovig et informez immédiatement votre médecin ou appelez le médecin d'urgence si vous remarquez de tels signes. Votre médecin initiera un traitement approprié.
Des cas de constipation accompagnée de graves complications ont été rapportés. Bon nombre de ces cas concernaient des patients ayant des antécédents de constipation ou qui utilisaient en même temps des médicaments susceptibles de provoquer une constipation. Dans certains cas graves, une hospitalisation a été nécessaire. Veuillez donc informer votre médecin si vous souffrez de constipation.
Les effets secondaires mentionnés ci-dessous peuvent également survenir. Si ces effets secondaires s'aggravent, veuillez en informer votre médecin ou votre pharmacien.
Fréquent (concerne 1 à 10 utilisateurs sur 100)
- Signes et symptômes de douleurs, rougeur, gonflement, démangeaisons au site d'injection (réactions au site d'injection);
- constipation;
- crampes musculaires;
- démangeaisons.
Si vous remarquez des effets secondaires, veuillez en informer votre médecin ou votre pharmacien. Ceci vaut en particulier pour les effets secondaires non mentionnés dans cette notice d'emballage.
À quoi faut-il encore faire attention?
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques concernant le stockage
Conserver au réfrigérateur (2–8 °C).
Conserver le stylo prérempli dans l'emballage d'origine pour le protéger de la lumière.
Conserver hors de portée des enfants.
Après avoir été sorti du réfrigérateur, Aimovig peut être conservé à température ambiante (15–25 °C) et doit ensuite être utilisé dans un délai de 14 jours. Aimovig ne doit pas être conservé à nouveau au réfrigérateur après avoir été mis à température ambiante.
N'utilisez pas ce médicament si le liquide contient des particules solides bien visibles, s'il est trouble ou nettement jaune.
Demandez à votre médecin ou à votre pharmacien comment éliminer le médicament lorsque vous ne l'utilisez plus.
Pour de plus amples renseignements, consultez votre médecin ou votre pharmacien, qui disposent d'une information détaillée destinée aux professionnels.
Que contient Aimovig?
Principes actifs
Chaque stylo prérempli de 1 ml contient 70 mg ou 140 mg d'érénumab.
Excipients
Saccharose, polysorbate 80, hydroxyde de sodium, acide acétique 99%, eau pour injection.
Numéro d’autorisation
66748 (Swissmedic)
Où obtenez-vous Aimovig? Quels sont les emballages à disposition sur le marché?
En pharmacie, sur ordonnance médicale.
Emballages de 1 stylo prérempli à 70 mg ou 140 mg.
Titulaire de l’autorisation
Novartis Pharma Schweiz AG, Risch; domicile: 6343 Rotkreuz
Cette notice d'emballage a été vérifiée pour la dernière fois en août 2020 par l'autorité de contrôle des médicaments (Swissmedic).
Notice d’utilisation pour Aimovig 70 mg/ml et 140 mg/ml solution injectable en stylo prérempli
Veuillez lire l’intégralité de ces instructions avant l’utilisation d’un stylo prérempli Aimovig. Il est important que vous ne tentiez pas de procéder vous-même à l’injection sans avoir reçu auparavant une formation de votre médecin, de votre pharmacien ou d’un spécialiste médical.
Présentation du stylo Aimovig 70 mg (avec corps bleu clair, bouton de démarrage violet, capuchon blanc et manchon de sécurité vert)
Vue avant utilisation |
|
Présentation du stylo Aimovig 140 mg (avec corps bleu foncé, bouton de démarrage gris, capuchon orange et manchon de sécurité jaune) |
Vue avant utilisation |
|
Important: l’aiguille se trouve à l’intérieur du manchon de sécurité vert (stylo à 70 mg) ou jaune (stylo à 140 mg): |


| |
|
|
|
|
|
|
Dans tous les cas cités, vous devez utiliser un nouveau stylo prérempli. En cas de doute, veuillez contacter le spécialiste médical qui s’occupe de vous. | |
|
|
| |
|
|
|
Écarter pour aplanir Écartez la peau pour l’aplanir en la tirant avec votre pouce et vos autres doigts dans les directions opposées. Il doit en résulter une zone ferme d’une largeur d’environ cinq centimètres. OU Pincer Pincez fermement la peau entre le pouce et les autres doigts. Il doit en résulter une zone d’une largeur d’environ cinq centimètres. |
|
|
|
|
|
|
|
|
| |











Che cos’è Aimovig e quando si usa?
Aimovig contiene il principio attivo erenumab. Erenumab è un anticorpo monoclonale umano. Gli anticorpi monoclonali sono delle proteine che riconoscono determinate proteine nell'organismo e si legano ad esse. Aimovig appartiene a un gruppo di medicamenti impiegati come anticorpi diretti contro il recettore del CGRP (peptide correlato al gene della calcitonina).
Aimovig agisce bloccando l'attività della molecola CGRP, la quale è stata messa in relazione con l'emicrania. Aimovig viene utilizzato per il trattamento dell'emicrania negli adulti per i quali è indicata una terapia preventiva. Aimovig viene utilizzato su prescrizione medica.
Quando non si può assumere/usare Aimovig?
Aimovig non deve essere usato in caso di allergia a erenumab o a una delle sostanze ausiliarie contenute in Aimovig (elencate nella sezione «Cosa contiene Aimovig?»).
L'uso di Aimovig non è stato studiato nei bambini e negli adolescenti di età inferiore a 18 anni. Non è noto se Aimovig sia sicuro ed efficace nei bambini e negli adolescenti. Per questo motivo, Aimovig non deve essere somministrato in questa fascia di età.
Quando è richiesta prudenza nella somministrazione/nell’uso di Aimovig?
Se manifesta sintomi di una grave reazione allergica come ad es. eruzione cutanea (prurito intenso alla cute con eruzione rossastra o pomfi in rilievo, la cosiddetta orticaria) o gonfiori, che in genere interessano il viso, la bocca, la lingua o la gola, o disturbi respiratori, sospenda immediatamente l'assunzione di Aimovig e informi subito il suo medico oppure contatti il medico del pronto soccorso.
Le reazioni allergiche gravi possono presentarsi entro pochi minuti, ma alcune possono tuttavia manifestarsi anche dopo più di una settimana dall'assunzione di Aimovig.
Durante il trattamento con Aimovig si può verificare costipazione con gravi complicazioni. Informi il medico se soffre di costipazione, ha sofferto di costipazione in passato o se assume in concomitanza medicamenti che possono causare costipazione. Il medico la monitorerà in caso di segni di costipazione grave e la tratterà di conseguenza.
Il cappuccio rimovibile della penna preriempita di Aimovig contiene lattice di gomma naturale essiccato, che può causare reazioni allergiche nelle persone sensibili al lattice. Informi il suo medico se è allergico al lattice.
Finora non vi sono indicazioni che l'assunzione di Aimovig comprometta la capacità di condurre veicoli e utilizzare macchine.
Questo medicamento (penna preriempita da 70 mg e da 140 mg) contiene meno di 1 mmol di sodio (23 mg) per penna preriempita, il che lo rende essenzialmente «privo di sodio».
Informi il suo medico o il suo farmacista, nel caso in cui
- soffre di altre malattie
- soffre di allergie o
- assume altri medicamenti (anche se acquistati di sua iniziativa).
Si può assumere/usare Aimovig durante la gravidanza o l’allattamento?
Aimovig non è stato valutato nelle donne in gravidanza. Non è noto se Aimovig possa causare effetti dannosi per il feto e, pertanto, Aimovig non dovrebbe essere usato durante la gravidanza. Informi il suo medico se pensa di essere incinta o intende iniziare una gravidanza o se sta allattando mentre sta utilizzando Aimovig. Non è noto se Aimovig passi nel latte materno. Molti medicamenti, fra cui gli anticorpi, vengono escreti nel latte materno, pertanto non si possono escludere rischi per i neonati/lattanti. È importante che informi il medico se sta allattando o prevede di allattare. Il medico deciderà se lei debba interrompere l'allattamento o piuttosto l'assunzione di Aimovig.
Come usare Aimovig?
L'indicazione alla terapia deve essere stabilita da un medico con esperienza nel trattamento dell'emicrania. Il medico la seguirà nelle successive fasi del trattamento.
Aimovig viene somministrato come iniezione sotto la pelle («iniezione sottocutanea»).
Insieme al suo medico, stabilirà se potrà praticare le iniezioni di Aimovig da solo/a.
È importante che non cerchi di praticare l'iniezione prima di aver ricevuto un'adeguata formazione dal medico o farmacista oppure da un operatore sanitario.
Per informazioni sulla corretta preparazione e somministrazione delle iniezioni di Aimovig a casa, consulti le istruzioni dettagliate per l'uso riportate in fondo a questo foglietto illustrativo.
Posologia di Aimovig
La dose di Aimovig è di 70 mg una volta al mese.
Nei pazienti che dopo alcuni mesi mostrano una risposta inadeguata a questo dosaggio, il medico può aumentare il dosaggio a 140 mg una volta al mese, se si ottiene una maggiore efficacia.
Non modifichi di propria iniziativa la posologia prescritta. Se ritiene che l'azione del medicamento sia troppo debole o troppo forte ne parli al suo medico o al suo farmacista.
Per quanto tempo si utilizza Aimovig?
Usi Aimovig una volta al mese per il periodo di tempo prescritto dal suo medico.
In caso di domande sulla durata dell'utilizzo di Aimovig, si rivolga al medico.
Se ha utilizzato una dose di Aimovig superiore a quella prescritta
Se ha utilizzato una dose di Aimovig superiore a quella prescritta si rivolga al medico.
Se ha dimenticato di utilizzare Aimovig
Se ha dimenticato di utilizzare Aimovig, recuperi il prima possibile. Quindi contatti il suo medico, che le dirà quando somministrare le dosi successive. Si attenga esattamente al piano di somministrazione stabilito dal medico.
Se interrompe l'uso di Aimovig
Non interrompa l'uso di Aimovig senza aver prima consultato il suo medico. Se interrompe il trattamento, i sintomi accusati potrebbero manifestarsi nuovamente.
Consulti il medico se ha domande sull'uso di Aimovig.
Bambini e adolescenti (sotto i 18 anni)
L'uso di Aimovig nei bambini e negli adolescenti sotto i 18 anni finora non è stato studiato. Non è noto se Aimovig sia sicuro ed efficace nei bambini e negli adolescenti. Per questo motivo, Aimovig non deve essere somministrato in questa fascia di età.
Quali effetti collaterali può avere Aimovig?
Possono manifestarsi reazioni allergiche gravi come ad es. eruzione cutanea (prurito intenso alla cute con eruzione rossastra o pomfi in rilievo, la cosiddetta orticaria) o gonfiori, che in genere interessano il viso, la bocca, la lingua o la gola, o disturbi respiratori. Se nota la comparsa di tali segni, sospenda immediatamente l'assunzione di Aimovig e informi subito il suo medico oppure contatti il medico del pronto soccorso. Il medico le prescriverà una terapia idonea.
Sono stati riportati casi di costipazione con gravi complicazioni. Molti di questi casi sono stati segnalati in pazienti che presentavano costipazione nell'anamnesi o che hanno assunto in concomitanza medicamenti che possono causare costipazione. Per alcuni casi gravi è stata necessaria l'ospedalizzazione. Informi il suo medico se soffre di costipazione.
Possono inoltre verificarsi gli effetti collaterali elencati di seguito. Se questi effetti collaterali peggiorano, informi il suo medico o il farmacista.
Comune (riguarda da 1 a 10 utilizzatori su 100)
- Segni e sintomi di dolore, arrossamento, gonfiore, prurito nella sede di iniezione (reazioni nella sede di iniezione)
- Costipazione
- Crampi muscolari
- Prurito
Se osserva effetti collaterali, si rivolga al suo medico, farmacista, soprattutto se si tratta di effetti collaterali non descritti in questo foglietto illustrativo.
Di che altro occorre tener conto?
Il medicamento non dev'essere utilizzato oltre la data indicata con «EXP» sul contenitore.
Indicazione di stoccaggio
Conservare in frigorifero (2–8 °C).
Conservare la penna preriempita nella confezione originale per proteggere il contenuto dalla luce.
Conservare fuori dalla portata dei bambini.
Una volta estratto dal frigorifero, Aimovig può essere conservato a temperatura ambiente (15–25 °C) e deve essere utilizzato entro 14 giorni. Una volta raggiunta la temperatura ambiente, Aimovig non deve essere rimesso in frigorifero.
Non utilizzare il medicamento se il liquido contiene particelle ben riconoscibili o appare torbido o inequivocabilmente giallo.
Chieda al suo medico o al farmacista come smaltire il medicamento quando non lo utilizzerà più.
Il medico o il farmacista, che sono in possesso di un'informazione professionale dettagliata, possono darle ulteriori informazioni.
Cosa contiene Aimovig?
Principi attivi
Ogni penna preriempita da 1 ml contiene 70 mg o 140 mg di erenumab.
Sostanze ausiliarie
Saccarosio, polisorbato 80, idrossido di sodio, acido acetico 99%, acqua per preparazioni iniettabili.
Numero dell’omologazione
66 748 (Swissmedic)
Dove è ottenibile Aimovig? Quali confezioni sono disponibili?
In farmacia, dietro presentazione della prescrizione medica.
Confezioni da 1 penna preriempita da 70 mg o 140 mg.
Titolare dell’omologazione
Novartis Pharma Schweiz AG, Risch; Domicilio: 6343 Rotkreuz
Questo foglietto illustrativo è stato controllato l'ultima volta nell'agosto 2020 dall'autorità competente in materia di medicamenti (Swissmedic).
Istruzioni per la preparazione di Aimovig soluzione iniettabile 70 mg/ml e 140 mg/ml in penna preriempita
Leggere interamente le presenti istruzioni prima di usare una penna preriempita di Aimovig. È importante che non cerchi di praticare l’iniezione prima di aver ricevuto un’adeguata formazione dal medico o farmacista oppure da un operatore sanitario.
Immagine della penna di Aimovig da 70 mg (con corpo blu chiaro, pulsante d’avvio viola, cappuccio bianco e dispositivo di sicurezza verde)
Panoramica prima della somministrazione |
|
Immagine della penna di Aimovig da 140 mg (con corpo blu scuro, pulsante d’avvio grigio, cappuccio arancione e dispositivo di sicurezza giallo) |
Panoramica prima della somministrazione |
|
Importante: l’ago si trova all’interno del dispositivo di sicurezza verde (penna da 70 mg) o giallo (penna da 140 mg): |


| |
|
|
|
|
|
|
In tutti questi casi si deve utilizzare una nuova penna preriempita. In caso di dubbi, rivolgersi all’operatore sanitario competente. | |
|
|
| |
|
|
|
Tendere la pelle Tendere la pelle tirandola con il pollice e le altre dita in direzioni opposte formando un’area tesa larga circa cinque centimetri. OPPURE Pizzicare Pizzicare la pelle saldamente tra il pollice e le dita, formando una piega larga circa cinque centimetri. |
|
|
|
|
|
|
|
|
| |










Zusammensetzung
Aimovig® 70 mg/ml, 140 mg/ml
Wirkstoffe
Erenumabum (gentechnisch hergestellt unter Verwendung von Ovarienzellen chinesischer Hamster).
Hilfsstoffe
Saccharum, Polysorbatum 80, Natrii hydroxidum corresp. Natrium 0,2 mg/ml, Acidum aceticum glaciale, Aqua ad iniectabilia
Darreichungsform und Wirkstoffmenge pro Einheit
Injektionslösung in einer Fertigspritze bzw. im Fertigpen zur subkutanen Anwendung:
1 Fertigspritze bzw. 1 Fertigpen zu 1 ml enthält 70 mg Erenumab in 1 ml (70 mg/ml) Injektionslösung
1 Fertigspritze bzw. 1 Fertigpen zu 1 ml enthält 140 mg Erenumab in 1 ml (140 mg/ml) Injektionslösung
Indikationen/Anwendungsmöglichkeiten
Prophylaktische Behandlung der Migräne bei Erwachsenen, sofern diese indiziert ist.
Dosierung/Anwendung
Die Indikation für die Therapie muss durch einen Arzt oder eine Ärztin mit Erfahrung auf dem Gebiet der Migränebehandlung gestellt und durch diese in der weiteren Behandlung begleitet werden. Bei mangelndem Therapieansprechen, beziehungsweise nach spätestens 12 Monaten sollte eine Reevaluation zur Fortführung der Therapie vorgenommen werden.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Empfohlen wird eine Aimovig-Dosis von 70 mg als subkutane Injektion einmal monatlich.
Bei Patienten, die auf diese Dosierung eine ungenügende Wirkung zeigen, kann die Dosierung auf 140 mg einmal monatlich gesteigert werden, solange dadurch eine bessere Wirkung nachweisbar ist.
Patienten mit Leberfunktionsstörungen
Es sind keine klinischen Studien bei Patienten mit einer Leberfunktionsstörung durchgeführt worden. Bei Patienten mit Leberinsuffizienz wird keine Dosisanpassung empfohlen, da Erenumab, ein humanes Immunglobulin G, nicht von Cytochrom P450-Enzymen metabolisiert wird und seine Clearance nicht in wesentlichem Umfang über die Leber erfolgt
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit leichter bis mittelschwerer Nierenfunktionsstörung ist keine Dosisanpassung erforderlich. Die pharmakokinetische Populationsanalyse integrierter Daten aus den klinischen Studien mit Aimovig ergaben keinen Unterschied zwischen Patienten mit leichter oder mittelschwerer Funktionsstörung und solchen mit normaler Nierenfunktion in Bezug auf die Pharmakokinetik von Erenumab. Patienten mit schwerer Nierenfunktionsstörung (eGFR <30 ml/min/1,73 m2) wurden nicht untersucht.
Ältere Patienten
In klinischen Studien wurden nicht genügend Patienten im Alter ab 65 Jahren erfasst, um eine Aussage darüber zu treffen, ob solche Patienten anders reagieren als jüngere Patienten. Eine Dosisanpassung ist nicht erforderlich, da die Pharmakokinetik von Erenumab nicht vom Alter beeinflusst wird.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Aimovig bei Kindern und Jugendlichen ist nicht untersucht worden. Aimovig darf daher in dieser Altersgruppe nicht angewendet werden.
Verspätete Dosisgabe
Wenn eine geplante Dosis vergessen wird, sollte sie so schnell wie möglich verabreicht werden, und weitere Injektionen sollten monatlich ab dem Datum der letzten Injektion geplant werden.
Art der Anwendung
Aimovig ist für die Selbstverabreichung in die Bauchdecke oder in den Oberschenkel durch den Patienten bestimmt. Die Injektionsstellen sollten gewechselt werden, und es sollte nicht in Stellen mit empfindlicher, verletzter, geröteter oder verhärteter Haut injiziert werden. Die Injektionen können auch in den Oberarm durch eine andere Person verabreicht werden.
Die Person, die das Präparat verabreicht, sollte durch eine Fachperson in die Verabreichungstechnik eingewiesen worden sein. Ausführliche Hinweise zur Anwendung sind der Patienteninformation (Packungsbeilage) zu entnehmen.
Kontraindikationen
Überempfindlichkeit gegenüber dem Wirkstoff Erenumab oder einem der Hilfsstoffe gemäss Zusammensetzung.
Warnhinweise und Vorsichtsmassnahmen
Überempfindlichkeitsreaktionen
Schwerwiegende Überempfindlichkeitsreaktionen, einschliesslich Ausschlag, Angioödem und anaphylaktoider Reaktionen, wurden mit Aimovig seit der Markteinführung berichtet. Diese Reaktionen können innerhalb von Minuten auftreten, einige können jedoch auch erst mehr als eine Woche nach der Behandlung in Erscheinung treten. Wenn eine schwere oder schwerwiegende Überempfindlichkeitsreaktion auftritt, muss die Anwendung von Aimovig sofort beendet werden und eine entsprechende Behandlung eingeleitet werden.
Obstipation mit schwerwiegenden Komplikationen:
Unter Aimovig kann es zu Obstipation mit schweren Komplikationen kommen (siehe «Unerwünschte Wirkungen»).
Patienten, die Aimovig einnehmen, sollten daher auf Anzeichen schwerer Obstipation überwacht und klinisch angemessen behandelt werden.
Die gleichzeitige Einnahme von Arzneimitteln, die mit einer verminderten gastrointestinalen Motilität assoziiert sind, kann das Risiko für schwere Obstipation und potenzielle Komplikationen erhöhen.
Patienten mit schweren Herz-Kreislauf-Erkrankungen
Patienten mit bestimmten schweren Herz-Kreislauf-Erkrankungen wurden von der Teilnahme an den klinischen Studien ausgeschlossen. Es liegen für diese Patienten keine Sicherheitsdaten vor.
Latex-empfindliche Patienten
Die abnehmbare Kappe der Aimovig-Fertigspritze und des Aimovig-Fertigpens enthält trockenen Naturkautschuklatex, der bei Personen mit Latexempfindlichkeit allergische Reaktionen hervorrufen kann.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Fertigspritze bzw. Fertigpen (70 mg/ml, 140 mg/ml), d.h. es ist nahezu «natriumfrei».
Interaktionen
In einer unverblindeten Studie zu pharmakokinetischen Arzneimittelwechselwirkungen zwischen Aimovig und einem oralen Kombinationspräparat zur Empfängnisverhütung bei gesunden weiblichen Probanden hatte Erenumab (140 mg subkutan [s. c.], Einzeldosis) keinen Einfluss auf die Pharmakokinetik eines oralen Kombinationspräparats zur Empfängnisverhütung, das Ethinylestradiol und Norgestimat enthielt.
In einer randomisierten, doppelblinden, placebokontrollierten Studie bei gesunden Probanden hatte die gleichzeitige Verabreichung von Erenumab (140 mg intravenös [i. v.], Einzeldosis) und Sumatriptan im Vergleich zur alleinigen Gabe von Sumatriptan keinen Einfluss auf den Ruheblutdruck. Aimovig hatte keinen Einfluss auf die Pharmakokinetik von Sumatriptan.
Erenumab wird nicht von Cytochrom P450 Enzymen metabolisiert. Daher sind Interaktionen mit Begleitmedikamenten, bei denen es sich um Substrate, Induktoren oder Inhibitoren von Cytochrom P450-Enzymen handelt, unwahrscheinlich.
Schwangerschaft/Stillzeit
Schwangerschaft
Es liegen keine hinreichenden Daten zur Anwendung von Aimovig bei Schwangeren vor. Tierexperimentelle Studien zeigten keine direkten oder indirekten schädlichen Auswirkungen in Bezug auf Reproduktionstoxizität (siehe «Präklinische Daten»).
Tierexperimentelle Studien sind nicht immer prädiktiv für die Reaktion beim Menschen, daher ist nicht bekannt, ob Aimovig für den Fötus schädlich ist, wenn es einer schwangeren Frau verabreicht wird. Während einer Schwangerschaft sollte Aimovig nicht angewendet werden, es sei denn dies ist eindeutig erforderlich.
Stillzeit
Es ist nicht bekannt, ob Aimovig in die menschliche Muttermilch übergeht. Da viele Arzneimittel, darunter Antikörper, in die Muttermilch ausgeschieden werden, kann ein Risiko für das Neugeborene/Kleinkind nicht ausgeschlossen werden. Daher sollte eine Entscheidung darüber getroffen werden, ob abgestillt oder Aimovig abgesetzt wird, wobei der mögliche Nutzen von Aimovig für die Mutter und der mögliche Nutzen des Stillens für den Säugling gegeneinander abzuwägen sind.
Fertilität
Es liegen keine Daten zu der Wirkung von Aimovig auf die Fertilität beim Menschen vor. Tierexperimentelle Studien zeigten keine Hinweise auf schädliche Wirkungen in Bezug auf die Fertilität auf (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Es wurden keine entsprechenden Studien durchgeführt. Aufgrund der verfügbaren Daten wird jedoch erwartet, dass Aimovig keinen Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen, hat.
Unerwünschte Wirkungen
Zur Beurteilung der Sicherheit von Aimovig im Vergleich zu einem Placebo bis zu 12 Wochen nach Behandlungsbeginn wurden Daten aus zwei klinischen Phase-3-Studien und zwei klinischen Phase-2-Studien bei Migräne kombiniert.
An den Placebo-kontrollierten Studien nahmen insgesamt 2656 Patienten teil. 1613 Patienten erhielten Aimovig und 1043 Patienten erhielten Placebo. Davon erhielten 893 Patienten eine Dosis von 70 mg Aimovig und 507 Patienten erhielten eine Dosis von 140 mg Aimovig.
Das Gesamtkollektiv zur Beurteilung der Sicherheit, einschliesslich der Patienten in der noch laufenden unverblindeten Verlängerungsphase mit Aimovig, bestand aus >2500 Patienten (>2300 Patientenjahre), die mindestens eine Dosis von Aimovig erhalten hatten: >2000 Patienten wurden mindestens 6 Monate behandelt und >1200 Patienten wurden mindestens 12 Monate behandelt.
Die Häufigkeiten werden wie folgt definiert:
«Sehr häufig» (≥1/10), «häufig» (<1/10, ≥1/100), «gelegentlich» (<1/100, ≥1/1'000), «selten» (<1/1'000, >1/10'000), «sehr selten» (<1/10'000).
Erkrankungen des Immunsystems
Häufigkeit unbekannt: Überempfindlichkeitsreaktionen, einschliesslich Ausschlag, Angioödem und anaphylaktoider Reaktionen (siehe «Warnhinweise und Vorsichtsmassnahmen.
Erkrankungen des Gastrointestinaltrakts
Häufig: Obstipation (siehe «Warnhinweise und Vorsichtsmassnahmen» «und «Beschreibung ausgewählter Nebenwirkungen»).
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Juckreiz.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Muskelkrämpfe.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Reaktionen an der Injektionsstelle (Schmerzen, Erythem oder Juckreiz).
Unerwünschte Wirkungen nach Markteinführung
Erkrankungen des Immunsystems
Häufigkeit unbekannt: Überempfindlichkeitsreaktionen, einschliesslich Ausschlag, Angioödem und anaphylaktoider Reaktionen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Erkrankung des Magen-Darm- Trakts
Häufigkeit unbekannt: Es wurde über Fälle von Obstipation mit schweren Komplikationen berichtet (siehe unter «Warnhinweise und Vorsichtsmassnahmen» und «Beschreibung ausgewählter Nebenwirkungen».
Beschreibung ausgewählter Nebenwirkungen
Immunogenität
In den 4 klinischen Studien (20120178, 20120295, 20120296 und 20120297) betrug die Inzidenz der Entwicklung von Antikörpern gegen Erenumab während der doppelblinden Behandlungsphase 6.3% (56/884) bei Patienten unter Behandlung mit 70 mg Erenumab (wovon 3 in-vitro neutralisierende Aktivität nachwiesen) und 2.6% (13/504) bei Patienten unter Behandlung mit 140 mg Erenumab (wovon in keinem Fall in-vitro neutralisierende Aktivität nachgewiesen wurde). Die mittleren Talspiegel von Erenumab in Woche 12 waren bei Anti-Erenumab-Antikörper-positiven Patienten 40% niedriger als bei antikörpernegativen Patienten. Die Entwicklung von Antikörpern gegen Erenumab hatte keine Auswirkungen auf die Wirksamkeit oder Sicherheit von Erenumab.
Die Inzidenz von Anti-Drug-Antikörpern (ADAs) hängt in hohem Mass von der Empfindlichkeit und Spezifität des Tests ab. Darüber hinaus kann die beobachtete Inzidenz der Antikörperpositivität (einschliesslich neutralisierender Antikörper) in einem Test von mehreren Faktoren beeinflusst werden, beispielsweise von der Testmethodik, der Probenhandhabung, dem Zeitpunkt der Probengewinnung, den Begleitmedikamenten und der Grundkrankheit. Aus diesem Grund kann der Vergleich der Inzidenz von Antikörpern gegen Erenumab mit der Inzidenz von Antikörpern gegen andere Arzneimittel irreführend sein.
Obstipation
Obstipation war eine der häufigsten Nebenwirkungen in klinischen Studien, über die während der 12-wöchigen placebokontrollierten Phase von klinischen Studien berichtet wurde (bis zu 3%).
Alle Fälle waren von leichtem oder mittlerem Schweregrad. Nach Markteinführung wurde zudem über Fälle von Obstipation mit schweren Komplikationen berichtet. Einige Fälle machten einen Spitalaufenthalt, einschliesslich chirurgischer Eingriffe notwendig. Bei den meisten Patienten trat die Obstipation nach der ersten Dosis von Aimovig auf, aber auch später im Behandlungsverlauf wurden Obstipationen berichtet. Die Therapie mit Aimovig wurde in den meisten gemeldeten Fällen mit schwerwiegender Obstipation unterbrochen. Viele dieser Fälle wurden bei Patienten gemeldet, die eine Obstipation in der Vorgeschichte hatten oder gleichzeitig Arzneimittel anwendeten, die mit einer verminderten gastrointestinalen Motilität assoziiert sind (siehe unter «Warnhinweise und Vorsichtsmassnahmen»).
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Es liegen keine Erfahrungen mit Überdosierungen von Aimovig in klinischen Studien vor. In klinischen Studien sind Dosen von bis zu 280 mg s. c. verabreicht worden, ohne dass eine dosisbegrenzende Toxizität festgestellt worden ist.
Im Falle einer Überdosierung sollte der/die Betroffene symptomatisch behandelt werden, und bei Bedarf sind unterstützende Massnahmen zu ergreifen.
Eigenschaften/Wirkungen
ATC-Code
N02CD01
Wirkungsmechanismus
Pharmakodynamik
Erenumab ist ein humaner monoklonaler antagonistischer Antikörper gegen den Calcitonin Gene Related Peptide Rezeptor (CGRP-Rezeptor) ohne wesentliche pharmakologische Wirkung auf Adrenomedullin-, Calcitonin- und Amylin-Rezeptoren und ohne agonistische Wirkung auf den CGRP-Rezeptor.
CGRP ist ein Neuropeptid, das die nozizeptive Signalübertragung moduliert und ein Vasodilatator, der mit der Pathophysiologie der Migräne in Verbindung gebracht worden ist.
Der CGRP-Rezeptor befindet sich an Orten, die für die Pathophysiologie der Migräne relevant sind. Erenumab konkurriert potent und spezifisch mit CGRP um die Bindung an den CGRP-Rezeptor und hemmt so die Funktion von CGRP am CGRP-Rezeptor.
In einer randomisierten, doppelblinden, placebokontrollierten Studie (20140254) zur Beurteilung der Wirksamkeit von Aimovig (140 mg i. v., Einzeldosis) bei Patienten mit stabiler Angina pectoris hatte Aimovig keine Verringerung der Belastungsdauer im Laufbandtest im Vergleich zu einem Placebo zur Folge und führte zu keiner Verschlimmerung der Myokardischämie.
Klinische Wirksamkeit
Chronische Migräne
Studie 1 (Studie 20120295)
Aimovig wurde in einer randomisierten, multizentrischen, 12‑wöchigen, placebokontrollierten, doppelblinden Studie zur Prophylaxe gegen chronische Migräne beurteilt. Insgesamt 667 Migränepatienten mit oder ohne Aura in der Vorgeschichte (≥15 Kopfschmerztage pro Monat mit ≥8 Migränetagen pro Monat) wurden randomisiert, um 12 Wochen lang alle 4 Wochen ein Placebo (n = 286), 70 mg Aimovig (n = 191) oder 140 mg Aimovig (n = 190) per subkutane Injektion zu erhalten.
Zum Studienbeginn betrug die mittlere Migränehäufigkeit ungefähr 18 Migränetage pro Monat und war in allen Behandlungsgruppen ähnlich. Den Patienten war es während der Studie gestattet, Akutbehandlungen gegen Kopfschmerzen anzuwenden, beispielsweise Triptane, Ergotamin-Derivate und NSARs.
Das mediane Alter der Patienten betrug 43 Jahre (Bereich: 18-66 Jahre), 83% waren weiblich und 94% waren weiss. Patienten mit vorbestehendem Myokardinfarkt, Schlaganfall, transitorischen ischämischen Attacken, instabiler Angina pectoris, koronarer arterieller Bypass-Operation oder anderen durchgeführten Revaskularisierungsverfahren innerhalb der letzten 12 Monaten vor dem Screening wurden aus Studie 1 ausgeschlossen.
Die primäre Outcome-Variable war die Veränderung der Migränetage pro Monat in Monat 3 gegenüber dem Studienbeginn. Zu den sekundären Outcome-Variablen zählten das Erreichen einer Verringerung der Migränetage pro Monat gegenüber dem Studienbeginn um 50 bis 100% (≥50% Responder) und die Veränderung der Anzahl der Tage mit migränespezifischer Akutmedikation pro Monat gegenüber dem Studienbeginn.
Abbildung 1 Veränderung der Anzahl der Migränetage pro Monat in Studie 1

Es sind die Kleinstquadrat-Mittelwerte und die 95%-Konfidenzintervalle angegeben.
Der p-Wert für die Differenz der Kleinstquadrat-Mittelwerte zwischen Erenumab und dem Placebo bei Beurteilung in Monat 3 (primäre Outcome-Variable) betrug <0,001.
Tabelle 1 Veränderungen der Wirksamkeit und den von den Patienten selbst berichteten
Outcome-Variablen in Woche 12 gegenüber dem Studienbeginn (Baseline) in Studie 1
Aimovig 70 mg (n = 188) | Aimovig 140 mg (n = 187) | Placebo (n = 281) | Behandlungs-unterschied/ Odds ratioc (95% KI) | p-Werta | |
|---|---|---|---|---|---|
≥50% MMT Responderc Prozent (%) | 39.9 | 41.2 | 23.5 | ORd 70 mg: 2.18 (1.46, 3.27) 140 mg: 2.34 (1.56; 3.51) | <0.001 |
Anzahl Tage mit migräne-spezifischer Akutmedikation (pro Monat)e Mittlere Veränderungb (95% KI) | -3.45 (-4.02, -2.87) | ‑4.13 (‑4.70; ‑3.56) | ‑1.58 (‑2.05; ‑1.11) | BU 70 mg: -1.86 (-2.60, -1.13) 140 mg: ‑2.55 (‑3.28; ‑1.82) | <0.001 |
BU = Behandlungsunterschied, KI = Konfidenzintervall; MMT = Migränetage pro Monat, OR = odds ratio a. Alle p‑Werte sind als nicht-angepasste p‑Werte angegeben, die nach Anpassung im Hinblick auf Mehrfachvergleiche statistisch signifikant sind. b. Die Veränderung der Kleinstquadrat-Mittelwerte gegenüber dem Studienbeginn in Monat 3, der Behandlungsunterschied und der p‑Wert beruhen auf einem linearen Gemische-Effekte-Modell unter Berücksichtigung der Behandlungsgruppe, des Werts pro Monat zum Studienbeginn, der Stratifizierungsfaktoren (Region [Nordamerika vs. Europa] und Medikationsübergebrauch [vorhanden vs. nicht vorhanden]), des geplanten Besuchs und der Interaktion zwischen Behandlungsgruppe und geplantem Besuch ohne Imputation bei fehlenden Daten. c. Responder sind definiert als Patienten, welche eine Verringerung von ≥50% in der Anzahl Migränetage pro Monat gegenüber Studienbeginn (Baseline) erreichen d. Das Odds-Ratio und der p‑Wert für ≥50% Responder in Monat 3 beruhen auf einem stratifizierten Cochran-Mantel‑Haenszel-Test nach Imputation fehlender Daten als Nicht-Ansprechen (Non‑Response). e. Migräne‑spezifische Medikationen sind beispielsweise Triptane und Ergotamin-Derivate. | |||||
Episodische Migräne
Studie 2 (Studie 20120296, STRIVE)
Bei Studie 2 handelte es sich um eine randomisierte, multizentrische, 24-wöchige, placebokontrollierte, doppelblinde Studie zur Beurteilung von Aimovig zur Prophylaxe gegen episodische Migräne. Insgesamt 955 Patienten mit Migräne mit einer Dauer von ≥12 Monaten mit oder ohne Aura und 4-14 Migränetagen pro Monat in der Krankengeschichte wurden randomisiert, um alle 4 Wochen während 24 Wochen mit 70 mg Aimovig (n=317), 140 mg Aimovig (n = 319) oder ein Placebo (n = 319) durch subkutane Injektion zu erhalten.
Zum Studienbeginn betrug die mittlere Migränehäufigkeit ungefähr 8 Migränetage pro Monat und war in allen Behandlungsgruppen ähnlich. Den Patienten war es während der Studie gestattet, Akutbehandlungen gegen Kopfschmerzen anzuwenden, beispielsweise Triptane, Ergotamin-Derivate und NSARs.
Das mediane Alter der Patienten betrug 42 Jahre (18-65 Jahre), 85% waren weiblich und 89% waren weiss. Patienten mit Medikationsübergebrauch, Patienten mit vorbestehendem Myokardinfarkt, Schlaganfall, transitorischen ischämischen Attacken, instabiler Angina pectoris, koronarer arterieller Bypass-Operation oder anderen durchgeführten Revaskularisierungsverfahren innerhalb der letzten 12 Monaten vor dem Screening wurden aus Studie 2 ausgeschlossen.
Der primäre Endpunkt war die Veränderung der Migränetage pro Monat in den Monaten 4-6 gegenüber dem Studienbeginn. Sekundäre Endpunkte waren unter anderem das Erreichen einer Verringerung der mittleren Migränetage pro Monat gegenüber dem Studienbeginn um 50 bis 100% (≥50% Responder) und die Veränderung der mittleren Anzahl der Tage mit migränespezifischer Akutmedikation pro Monat gegenüber dem Studienbeginn.
Unterschiede zum Placebo waren bereits nach einem Monat feststellbar.
Abbildung 2 Veränderung der Anzahl der Migränetage pro Monat in Studie 2a

a Es sind die Kleinstquadrat-Mittelwerte und die 95%-Konfidenzintervalle angegeben.
Der p-Wert für die Differenz der Kleinstquadrat-Mittelwerte zwischen Erenumab und dem Placebo als Durchschnittswert der Monate 4, 5 und 6 (primäre Outcome-Variable) betrug <0,001
Tabelle 2 Veränderungen der Wirksamkeit und den von den Patienten selbst berichteten
Outcome-Variablen gegenüber dem Studienbeginn (Baseline) in den Wochen 13-24 in Studie 2
Aimovig 70 mg (n = 312) | Aimovig 140 mg (n = 318) | Placebo (n = 316) | Behandlungsunterschied/Odds Ratio (95% CI) | p-Werta | |
|---|---|---|---|---|---|
≥50% MMT Responderc Prozent (%) | 43.3 | 50.0 | 26.6 | ORd 70 mg: 2.13 (1.52, 2.98) 140 mg: 2.81 (2.01, 3.94) | <0.001 |
Anzahl der Tage mit migränespezifischer Akutmedikation pro Monate Mittlere Veränderungb (95% KI) | -1.13 (-1.34, -0.92) | ‑1.61 (‑1.83; ‑1.40) | ‑0.20 (‑0.41; 0.02) | BU 70 mg: -0.94 (-1.23, -0.64) 140 mg: ‑1.42 (‑1.71; ‑1.12) | <0.001 |
BU = Behandlungsunterschied; KI = Konfidenzintervall; MMT = Migränetage pro Monat; OR = odds ratio a Alle p Werte sind als nicht-angepasste p Werte angegeben, die nach Anpassung im Hinblick auf Mehrfachvergleiche statistisch signifikant sind. b Die Veränderung der Kleinstquadrat-Mittelwerte gegenüber dem Studienbeginn in den Monaten 4-6, der Behandlungsunterschied und der p-Wert beruhen auf einem linearen Gemischte Effekte-Modell unter Berücksichtigung der Behandlungsgruppe, des Werts zum Studienbeginn, der Stratifizierungsfaktoren (Region [Nordamerika vs. Rest der Welt] und vorgängige Anwendung prophylaktischer Medikamente [keine Anwendung, nur vorgängige Anwendung, gleichzeitige Anwendung]), des geplanten Besuchs und der Interaktion zwischen Behandlungsgruppe und geplantem Besuch ohne Imputation bei fehlenden Daten. c Responder sind definiert als Patienten, welche eine Verringerung von ≥50% in der Anzahl Migränetage pro Monat gegenüber Studienbeginn (Baseline) erreichen d Das Odds-Ratio und der p-Wert für ≥50% Responder in den Monaten 4-6 beruhen auf einem stratifizierten Cochran-Mantel-Haenszel-Test nach Imputation fehlender Daten als Nicht-Ansprechen (Non-Response). e Migräne-spezifische Medikationen sind beispielsweise Triptane und Ergotamin-Derivate. | |||||
Pharmakokinetik
Absorption
Erenumab weist infolge der Bindung an den CGRP-Rezeptor eine nicht lineare Kinetik auf. Die subkutane Gabe einer 70-mg und 140-mg-Dosis an gesunde Probanden führte zu einem Cmax-Mittelwert (Standardabweichung [standard deviation, SD]) von 6.1 [2.1] und 15.8 [4.8] µg/ml und zu einem AUClast-Mittelwert [SD] von 159 [58] und 505 [139] Tag*µg/ml.
Die Serum-Talspiegel waren nach subkutaner Gabe von 70-mg und 140-mg-Dosen alle 4 Wochen um weniger als das Doppelte erhöht (Cmin [SD] 5.7 [3.1] und 6.2 [2.9] µg/ml unter 70-mg Dosierung und Cmin [SD] 12.8 [6.5] und 14.9 [6.5] µg/ml unter 140-mg Dosierung bei Patienten mit episodischer bzw. chronischer Migräne) und näherten sich nach 12-wöchiger Gabe dem Steady-State an.
Nach Gabe einer subkutanen Einzeldosis von 70 mg oder 140 mg Aimovig an gesunde Erwachsene betrug die mediane Zeit bis zur Serumspitzenkonzentration ungefähr 6 Tage. Die absolute Bioverfügbarkeit geschätzt anhand der pharmakokinetischen Populationsanalyse betrug 82%.
Distribution
Nach Gabe einer intravenösen Einzeldosis von 140 mg wurde das mittlere Distributionsvolumen (SD) während der terminalen Phase (Vz) auf 3.86 (0.77) Liter geschätzt.
Metabolismus
Die effektive Halbwertszeit von Aimovig geschätzt anhand der pharmakokinetischen Populationsanalyse beträgt 28 Tage.
Elimination
Bei Aimovig wurden zwei Eliminationsphasen festgestellt. Bei niedrigen Konzentrationen erfolgt die Elimination von Aimovig hauptsächlich durch sättigbare Bindung an das Zielmolekül (CGRP-R), bei höheren Konzentrationen weitgehend über einen unspezifischen, nicht sättigbaren proteolytischen Eliminationsweg.
Linearität/Nicht Linearität
Nach subkutaner, monatlicher Verabreichung von Dosen von 70 und 140 mg ist die Pharmakokinetik von Aimovig innerhalb des Dosierungsintervalls linear mit einer dosisproportionalen Erhöhung der Serumkonzentration.
Kinetik spezieller Patientengruppen
Die populationspharmakokinetische (PK) Analyse ergab, dass die Pharmakokinetik von Erenumab in keiner der zugelassenen Patientengruppen durch das Alter, das Geschlecht, die Ethnie, den Migräne-Subtyp (episodische oder chronische Migräne) oder die geschätzte glomeruläre Filtrationsrate (eGFR) beeinflusst wurde. Patienten mit schwerer Nierenfunktionsstörung (eGFR <30 ml/min/1,73 m2) wurden nicht untersucht.
Präklinische Daten
Auf der Grundlage herkömmlicher Studien der Sicherheitspharmakologie, der Toxizität bei Mehrfachdosierung und der reproduktions- und entwicklungsbezogenen Toxizität lassen die präklinischen Daten keine spezielle Gefahr für den Menschen erkennen.
Mutagenität
Das Mutagenitätspotential von Aimovig ist nicht untersucht worden. Es ist jedoch unwahrscheinlich, dass monoklonale Antikörper DNA oder Chromosomen verändern.
Karzinogenität
Es wurden keine Studien zur Karzinogenität mit Aimovig durchgeführt. Aimovig ist bei Nagern pharmakologisch nicht aktiv, weist aber bei Cynomolgus-Affen biologische Aktivität auf; diese Tierart ist jedoch kein geeignetes Modell zur Untersuchung des Risikos einer Tumorbildung.
Reproduktionstoxizität
In einer Reproduktionsstudie bei Cynomolgus-Affen (ePPND, erweiterte prä- und postnatale Entwicklungsstudie) wurden keine Auswirkungen auf die Trächtigkeit und auf die embryo-fötale oder postnatale Entwicklung (bis zu einem Alter von 6 Monaten) festgestellt, wenn Erenumab während der Trächtigkeit der Tiere in Dosen verabreicht wurde, die eine 17-mal höhere Exposition bewirkten als sie bei Patienten unter Behandlung mit 140 mg einmal monatlich erreicht wird (basierend auf dem AUC-Wert im Serum). Bei den neugeborenen Affen war Erenumab bei der Geburt im Serum nachweisbar, was bestätigt, dass Erenumab, wie andere IgG-Antikörper auch, die Plazentaschranke passiert.
Weitere Daten (Lokale Toxizität, Phototoxizität, Immunotoxizität)
In Studien zur chronischen Toxizität bei geschlechtsreifen Affen, denen bis zu 6 Monate lang zweimal wöchentlich eine Dosis von bis zu 150mg/kg Aimovig subkutan verabreicht worden war, wurden bei systemischen Expositionen, die bis zu 123 Mal höher waren als bei Gabe der klinischen Dosis von 140 mg alle 4 Wochen (basierend auf dem AUC-Wert im Serum), keine unerwünschten Wirkungen festgestellt. Ausserdem wurden in diesen Studien keine unerwünschten Wirkungen auf Surrogatmarker für die Fertilität (pathologisch-anatomische oder histopathologische Veränderungen in Fortpflanzungsorganen) festgestellt.
Sonstige Hinweise
Inkompatibilitäten
Da keine Kompatibilitätsstudien vorliegen, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden. Die Fertigspritze bzw. der Fertigpen sind nur für den Einmalgebrauch bestimmt.
Besondere Lagerungshinweise
Im Kühlschrank (2-8 °C) lagern.
Die Fertigspritze oder den Fertigpen in der Originalverpackung im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Nachdem Aimovig aus dem Kühlschrank genommen worden ist, kann es bei Raumtemperatur (15-25 °C) aufbewahrt werden und muss dann aber innerhalb von 14 Tagen angewendet werden. Aimovig darf nicht wieder im Kühlschrank aufbewahrt werden, nachdem es Raumtemperatur angenommen hat.
Hinweise für die Handhabung
Vor Gebrauch sollte die Flüssigkeit kontrolliert werden. Nicht injizieren, wenn die Flüssigkeit trüb oder deutlich gelb ist, oder wenn darin Flocken oder Partikel enthalten sind.
Vor der subkutanen Verabreichung Aimovig mindestens 30 Minuten bei Raumtemperatur stehen lassen.
Das Präparat nicht schütteln.
Zulassungsnummer
66620, 66748 (Swissmedic)
Zulassungsinhaberin
Novartis Pharma Schweiz AG. Risch; Domizil: 6343 Rotkreuz
Stand der Information
August 2020
Composizione
Aimovig® 70 mg/ml, 140 mg/ml
Principi attivi
Erenumab (prodotto mediante manipolazione genetica utilizzando cellule ovariche di criceto cinese).
Sostanze ausiliarie
Saccarosio, polisorbato 80, idrossido di sodio corrisp. a 0.2 mg/ml di sodio, acido acetico glaciale, acqua per preparazioni iniettabili.
Forma farmaceutica e quantità di principio attivo per unità
Soluzione iniettabile in una siringa preriempita o in penna preriempita per uso sottocutaneo:
1 siringa preriempita o 1 penna preriempita da 1 ml contiene 70 mg di erenumab in 1 ml (70 mg/ml) di soluzione iniettabile
1 siringa preriempita o 1 penna preriempita da 1 ml contiene 140 mg di erenumab in 1 ml (140 mg/ml) di soluzione iniettabile
Indicazioni/Possibilità d'impiego
Trattamento profilattico dell'emicrania negli adulti, se indicato.
Posologia/Impiego
L'indicazione alla terapia deve essere stabilita e seguita nelle fasi successive da un medico con esperienza nel trattamento dell'emicrania. In assenza di risposta alla terapia o al più tardi dopo 12 mesi, è necessaria una nuova valutazione per continuare la terapia.
Per garantire la tracciabilità dei medicamenti biotecnologici, si raccomanda di prendere nota del nome commerciale e del numero di lotto in occasione di ogni trattamento.
La dose raccomandata di Aimovig è di 70 mg per iniezione sottocutanea una volta al mese.
Nei pazienti che dopo alcuni mesi mostrano una risposta inadeguata a questo dosaggio, il medico può aumentare il dosaggio a 140 mg una volta al mese, se con questo si ottiene una maggiore efficacia.
Pazienti con disturbi della funzionalità epatica
Non sono stati effettuati studi clinici su pazienti con insufficienza epatica. Nei pazienti con insufficienza epatica non si consigliano aggiustamenti della dose, poiché erenumab, un'immunoglobulina umana G, non viene metabolizzato dagli enzimi del citocromo P450 e la sua clearance non avviene in misura significativa attraverso il fegato.
Pazienti con disturbi della funzionalità renale
Non è richiesto alcun aggiustamento della dose nei pazienti con insufficienza renale da lieve a moderata. L'analisi di popolazione farmacocinetica di dati integrati dagli studi clinici di Aimovig non ha mostrato differenze tra i pazienti con insufficienza lieve o moderata e quelli con funzionalità renale normale rispetto alla farmacocinetica di erenumab. I pazienti con insufficienza renale grave (eGFR <30 ml/min/1.73 m2) non sono stati valutati.
Pazienti anziani
Gli studi clinici non hanno coinvolto un numero di pazienti di età superiore ai 65 anni sufficiente per valutare se essi rispondano in modo diverso rispetto ai pazienti più giovani. L'aggiustamento della dose non è necessario poiché la farmacocinetica di erenumab non è influenzata dall'età.
Bambini e adolescenti
La sicurezza e l'efficacia di Aimovig nei bambini e negli adolescenti non sono state valutate. Per questo motivo, Aimovig non deve essere somministrato in questa fascia di età.
Somministrazione ritardata della dose
Se si dimentica una dose programmata, assumerla il prima possibile e programmare le iniezioni successive mensilmente dalla data dell'ultima iniezione.
Modo di somministrazione
Aimovig è destinato all'autosomministrazione nell'addome o nella coscia da parte del paziente. È necessario cambiare puntualmente le sedi di iniezione ed evitare di iniettare il medicamento in aree di pelle sensibile, danneggiata, arrossata o indurita. Le iniezioni possono anche essere praticate nel braccio da un'altra persona.
La persona che somministra il preparato deve essere stata istruita da un operatore sanitario specializzato nella tecnica di somministrazione. Per istruzioni dettagliate sulla modalità di somministrazione, fare riferimento all'informazione destinata ai pazienti (foglietto illustrativo).
Controindicazioni
Ipersensibilità al principio attivo erenumab o ad una qualsiasi delle sostanze ausiliarie secondo composizione.
Avvertenze e misure precauzionali
Reazioni di ipersensibilità
Dall'introduzione sul mercato, sono state riportate reazioni di ipersensibilità severe associate ad Aimovig, comprendenti eruzione cutanea, angioedema e reazioni anafilattoidi. Tali reazioni possono presentarsi entro pochi minuti, tuttavia alcune possono manifestarsi anche dopo più una settimana dal trattamento. In caso di reazione di ipersensibilità grave o severa, l'assunzione di Aimovig deve essere immediatamente sospesa e deve essere avviato un trattamento adeguato.
Stitichezza ostinata e complicazioni gravi
Durante il trattamento con Aimovig può verificarsi stitichezza ostinata con complicazioni gravi (vedere «Effetti indesiderati»).
I pazienti che assumono Aimovig devono essere monitorati per segni di stitichezza ostinata grave e clinicamente trattati in modo appropriato.
L'assunzione concomitante di medicamenti associati a una motilità gastrointestinale ridotta causa un aumento del rischio di stitichezza ostinata grave con potenziali complicazioni.
Pazienti con patologie cardiovascolari gravi
I pazienti con determinate patologie cardiovascolari gravi sono stati esclusi dalla partecipazione agli studi clinici. Non sono disponibili dati sulla sicurezza per questi pazienti.
Pazienti sensibili al lattice
Il cappuccio rimovibile della siringa preriempita di Aimovig e della penna preriempita di Aimovig contiene lattice di gomma naturale essiccato che può causare reazioni allergiche nelle persone sensibili al lattice.
Questo medicamento contiene meno di 1 mmol di sodio (23 mg) per siringa preriempita e penna preriempita (70 mg/ml, 140 mg/ml), il che lo rende essenzialmente «privo di sodio».
Interazioni
In uno studio in aperto sulle interazioni farmacocinetiche tra Aimovig e un preparato contraccettivo combinato orale in volontarie sane, erenumab (140 mg per via sottocutanea, [s. c.], dose singola) non ha avuto effetti sulla farmacocinetica di un preparato contraccettivo combinato orale contenente etinilestradiolo e norgestimato.
In uno studio randomizzato, in doppio cieco, controllato con placebo e condotto su volontari sani, la co-somministrazione di erenumab (140 mg per via endovenosa, [e. v.], dose singola) e sumatriptan non ha avuto alcun effetto sulla pressione arteriosa a riposo rispetto al solo sumatriptan. Aimovig non ha avuto alcun effetto sulla farmacocinetica di sumatriptan.
Erenumab non viene metabolizzato dagli enzimi del citocromo P450. Pertanto, le interazioni con medicamenti concomitanti, che sono substrati, induttori o inibitori degli enzimi del citocromo P450, sono improbabili.
Gravidanza/Allattamento
Gravidanza
Non sono disponibili dati sufficienti sull'utilizzo di Aimovig in gravidanza. Gli studi sugli animali non hanno mostrato effetti dannosi diretti o indiretti per quanto riguarda la tossicità per la riproduzione (vedere «Dati preclinici»).
Poiché gli studi sugli animali non sono sempre predittivi della risposta nell'uomo, non è noto se Aimovig sia dannoso per il feto se somministrato a una donna incinta. Non somministrare Aimovig durante la gravidanza, a meno che ciò non sia inequivocabilmente necessario.
Allattamento
Non è noto se Aimovig passi nel latte materno umano. Molti medicamenti, fra cui gli anticorpi, vengono escreti nel latte materno, pertanto non si possono escludere rischi per i neonati/lattanti. Di conseguenza, è necessario decidere se interrompere l'allattamento o sospendere Aimovig, considerando i potenziali benefici di Aimovig per la madre e i potenziali benefici dell'allattamento al seno per il bambino.
Fertilità
Non sono disponibili dati sugli effetti di Aimovig sulla fertilità nell'uomo. Negli studi sugli animali non sono stati riscontrati effetti dannosi sulla fertilità (vedere «Dati preclinici»).
Effetti sulla capacità di condurre veicoli e sull'impiego di macchine
Non sono stati effettuati studi in merito. Tuttavia, sulla base dei dati disponibili, Aimovig non dovrebbe avere alcun effetto sulla capacità di guidare veicoli o sulla capacità di utilizzare macchine.
Effetti indesiderati
Per valutare la sicurezza di Aimovig rispetto al placebo fino a 12 settimane dall'inizio del trattamento, sono stati combinati i dati di due studi clinici di fase 3 e di due studi clinici di fase 2 sull'emicrania.
Un totale di 2656 pazienti ha preso parte a studi controllati con placebo. 1613 pazienti hanno ricevuto Aimovig e 1043 pazienti hanno ricevuto placebo. Dei pazienti trattati con il farmaco, 893 pazienti hanno ricevuto una dose di 70 mg di Aimovig e 507 pazienti hanno ricevuto una dose di 140 mg di Aimovig.
La popolazione totale per la valutazione della sicurezza, compresi i pazienti nella fase di estensione in aperto di Aimovig in corso, era composta da >2500 pazienti (>2300 anni paziente) che hanno ricevuto almeno una dose di Aimovig: >2000 pazienti sono stati trattati per almeno 6 mesi e >1200 pazienti sono stati trattati per almeno 12 mesi.
Le frequenze sono definite come segue:
«molto comune» (≥1/10), «comune» (<1/10, ≥1/100), «non comune» (<1/100, ≥1/1000), «raro» (<1/1000, >1/10 000), «molto raro» (<1/10 000).
Disturbi del sistema immunitario
Frequenza non nota: reazioni di ipersensibilità, inclusi eruzione cutanea, angioedema e reazioni anafilattoidi (vedere «Avvertenze e misure precauzionali»).
Patologie gastrointestinali
Comune: stipsi (vedere «Avvertenze e misure precauzionali» e «Descrizione di alcuni effetti collaterali»).
Patologie della cute e del tessuto sottocutaneo
Comune: prurito.
Patologie del sistema muscoloscheletrico e del tessuto connettivo
Comune: crampi muscolari.
Patologie generali e condizioni relative alla sede di somministrazione
Comune: reazioni nella sede di iniezione (dolore, eritema o prurito).
Effetti indesiderati dopo l'introduzione sul mercato
Disturbi del sistema immunitario
Frequenza non nota: reazioni di ipersensibilità, inclusi reazione cutanea, angioedema e reazioni anafilattoidi (vedere «Avvertenze e misure precauzionali»).
Disturbi gastrointestinali
Frequenza non nota: sono stati riportati casi di stitichezza ostinata con complicazioni gravi (vedere «Avvertenze e misure precauzionali» e «Descrizione di alcuni effetti collaterali»).
Descrizione di alcuni effetti collaterali
Immunogenicità
Nei 4 studi clinici (20120178, 20120295, 20120296 e 20120297) l'incidenza dello sviluppo di anticorpi anti-erenumab durante la fase di trattamento in doppio cieco è stata del 6.3% (56/884) in pazienti trattati con erenumab 70 mg (di cui 3 hanno dimostrato attività neutralizzante in vitro) e del 2.6% (13/504) in pazienti trattati con erenumab 140 mg (di cui non è stata dimostrata alcuna attività neutralizzante in vitro). I livelli di valle medi di erenumab alla settimana 12 erano del 40% più bassi nei pazienti con anticorpi anti-erenumab rispetto ai pazienti anticorpo-negativi. Lo sviluppo di anticorpi anti-erenumab non ha avuto alcun impatto sull'efficacia o la sicurezza di erenumab.
L'incidenza di anticorpi anti-medicamento (ADA) dipende in larga misura dalla sensibilità e dalla specificità del test. Inoltre, l'incidenza osservata della positività anticorpale (compresi gli anticorpi neutralizzanti) in un test può essere influenzata da diversi fattori, tra cui ad es. la metodologia del test, la manipolazione del campione, i tempi di raccolta del campione, la somministrazione concomitante di altri medicamenti e la patologia di base. Per tale ragione, il confronto dell'incidenza degli anticorpi anti-erenumab con l'incidenza di anticorpi verso altri medicamenti può essere fuorviante.
Stitichezza ostinata
La stitichezza ostinata è stata uno degli effetti collaterali più comuni durante gli studi clinici, segnalati durante la fase di 12 settimane controllata con placebo degli studi clinici (fino al 3%).
Tutti i casi presentavano un grado di severità da lieve a moderato. Dopo l'introduzione sul mercato, sono stati riportati casi di stitichezza ostinata con complicazioni gravi. Alcuni casi hanno reso necessaria l'ospedalizzazione incluso il ricorso a interventi chirurgici. La maggior parte dei pazienti ha manifestato stitichezza ostinata dopo la prima dose di Aimovig, ma è stata riportata stitichezza ostinata anche in seguito nel corso del trattamento. La terapia con Aimovig è stata interrotta nella maggior parte dei casi segnalati di stitichezza ostinata grave. Molti di questi casi sono stati segnalati in pazienti che presentavano stitichezza ostinata nell'anamnesi o che hanno assunto in concomitanza medicamenti associati a una motilità gastrointestinale ridotta (vedere «Avvertenze e misure precauzionali»).
La notifica di effetti collaterali sospetti dopo l'omologazione del medicamento è molto importante. Consente una sorveglianza continua del rapporto rischio-benefico del medicamento. Chi esercita una professione sanitaria è invitato a segnalare qualsiasi nuovo o grave effetto collaterale sospetto attraverso il portale online ElViS (Electronic Vigilance System). Maggiori informazioni sul sito www.swissmedic.ch.
Posologia eccessiva
Durante gli studi clinici non sono stati riportati casi di sovradosaggio di Aimovig. Negli studi clinici sono state somministrate dosi per via sottocutanea fino a 280 mg senza osservare la comparsa di tossicità dose-limitante.
In caso di sovradosaggio, la persona interessata dovrebbe essere trattata sintomaticamente e, se necessario, devono essere adottate misure di supporto.
Proprietà/Effetti
Codice ATC
N02CD01
Meccanismo d'azione
Farmacodinamica
Erenumab è un anticorpo monoclonale umano antagonista del recettore del peptide correlato al gene della calcitonina (recettore del CGRP) senza attività farmacologica significativa sui recettori di adrenomedullina, calcitonina e amilina e senza effetto agonistico sul recettore del CGRP.
Il CGRP è un neuropeptide che modula il segnale nocicettivo ed è un vasodilatatore che è stato associato alla fisiopatologia dell'emicrania.
Il recettore CGRP si trova in siti rilevanti per la fisiopatologia dell'emicrania. Erenumab compete in modo potente e specifico con il CGRP per il legame al proprio recettore (CGRP), inibendone così la funzione a tale livello.
In uno studio randomizzato, in doppio cieco, controllato con placebo (20140254) per valutare l'efficacia di Aimovig (140 mg e. v., dose singola) in pazienti con angina pectoris stabile, Aimovig non ha ridotto la durata dell'esercizio fisico nel test sul tapis roulant rispetto al placebo e non ha determinato un peggioramento dell'ischemia miocardica.
Efficacia clinica
Emicrania cronica
Studio 1 (Studio 20120295)
Aimovig è stato valutato in uno studio randomizzato, multicentrico, di 12 settimane, controllato con placebo e in doppio cieco per la profilassi dell'emicrania cronica. Sono stati randomizzati in totale 667 pazienti con anamnesi di emicrania con o senza aura (≥15 giorni di cefalea al mese con ≥8 giorni di emicrania al mese) per ricevere un placebo (n = 286), 70 mg di Aimovig (n = 191) o 140 mg di Aimovig (n = 190) mediante iniezione sottocutanea ogni 4 settimane per 12 settimane.
Al basale, la frequenza media di emicrania era di circa 18 giorni di emicrania al mese ed era simile in tutti i gruppi di trattamento. Ai pazienti è stato permesso di usare trattamenti acuti per la cefalea durante lo studio, come i triptani, i derivati dell'ergotamina e i FANS.
L'età mediana dei pazienti era di 43 anni (range: da 18 a 66 anni), l'83% era di sesso femminile e il 94% era di razza bianca. I pazienti con preesistente infarto miocardico, ictus, attacchi ischemici transitori, angina pectoris instabile, intervento di bypass artero-coronarico o altre procedure di rivascolarizzazione eseguite nei 12 mesi precedenti allo screening, sono stati esclusi dalla partecipazione allo Studio 1.
La principale variabile di outcome era la variazione dei giorni di emicrania al mese del mese 3 rispetto al basale. Le variabili di outcome secondarie includevano una riduzione dal 50% al 100% dei giorni di emicrania al mese rispetto al basale (≥50% di responder) e una variazione nel numero di giorni con medicamenti acuti specifici per emicrania al mese rispetto al basale.
Figura 1 Variazione del numero di giorni di emicrania al mese nello Studio 1

Sono indicati la media dei minimi quadrati e gli intervalli di confidenza al 95%.
Il valore p della differenza nella media dei minimi quadrati tra erenumab e il placebo quando valutato al mese 3 (variabile di outcome primaria) era <0.001.
Tabella 1 Variazioni dell'efficacia e delle variabili di outcome riferite dal paziente alla settimana 12 rispetto all'inizio dello studio (basale) nello Studio 1
Aimovig 70 mg (n = 188) | Aimovig 140 mg (n = 187) | Placebo (n = 281) | Differenza di trattamento/ odds ratioc (IC 95%) | valore pa | |
|---|---|---|---|---|---|
Pazienti responder con ≥50% MMDc Percentuale (%) | 39.9 | 41.2 | 23.5 | ORd 70 mg: 2.18 140 mg: 2.34 | <0.001 |
Numero di giorni con medicamenti acuti specifici per emicrania (al mese)e Variazione mediab (IC 95%) | ‑3.45 (‑4.02; ‑2.87) | ‑4.13 (‑4.70; ‑3.56) | ‑1.58 (‑2.05; ‑1.11) | DT 70 mg: -1.86 140 mg: ‑2.55 | <0.001 |
DT = differenza di trattamento, IC = intervallo di confidenza; MMD = giorni di emicrania al mese, OR = odds ratio a. Tutti i valori p sono riportati come valori p non regolati, che sono statisticamente significativi dopo il raccordo per confronti multipli. b. La variazione dei minimi quadrati al basale al mese 3, la differenza di trattamento e il valore p si basano su un modello di effetti di miscela lineare, tenendo conto di gruppo di trattamento, valore al basale al mese, fattori di stratificazione (regione [Nord America vs. Europa] e uso eccessivo di medicamenti [presente vs. assente]), visita programmata e interazione tra gruppo di trattamento e visita programmata senza imputazione dei dati mancanti. c. I responder sono definiti come pazienti che raggiungono una riduzione ≥50% del numero di giorni di emicrania al mese rispetto all'inizio dello studio (basale). d. L'odd ratio e il valore p per ≥50% di responder al mese 3 sono basati su un test stratificato di Cochran-Mantel-Haenszel dopo l'imputazione dei dati mancanti come non risposta (non response). e. I medicamenti specifici per l'emicrania comprendono i triptani e i derivati dell'ergotamina. | |||||
Emicrania episodica
Studio 2 (Studio 20120296, STRIVE)
Lo Studio 2 è stato uno studio randomizzato, a 24 settimane, multicentrico, controllato con placebo, in doppio cieco, condotto per valutare Aimovig nella prevenzione dell'emicrania episodica. Sono stati randomizzati in totale 955 pazienti con emicrania di durata ≥12 mesi con o senza aura e anamnesi di 4–14 giorni di emicrania al mese per ricevere Aimovig 70 mg (n = 317), Aimovig 140 mg (n = 319) o un placebo (n = 319) per iniezione sottocutanea ogni 4 settimane per 24 settimane.
Al basale, la frequenza media di emicrania era di circa 8 giorni di emicrania al mese ed era simile in tutti i gruppi di trattamento. Ai pazienti è stato permesso di usare trattamenti acuti per la cefalea durante lo studio, come i triptani, i derivati dell'ergotamina e i FANS.
L'età mediana era di 42 anni (18–65 anni), l'85% era di sesso femminile e l'89% era di razza bianca. I pazienti con abuso di farmaci e i pazienti con preesistente infarto miocardico, ictus, attacchi ischemici transitori, angina pectoris instabile, intervento di bypass artero-coronarico o altre procedure di rivascolarizzazione eseguite nei 12 mesi precedenti allo screening sono stati esclusi dalla partecipazione allo Studio 2.
L'endpoint primario era la variazione dei giorni di emicrania al mese nei mesi 4–6 rispetto al basale. Gli endpoint secondari includevano tra gli altri una riduzione dal 50% al 100% dei giorni di emicrania medi al mese rispetto al basale (≥50% di responder) e una variazione nel numero medio di giorni con medicamenti acuti specifici per emicrania al mese rispetto al basale.
Le differenze con il placebo erano già evidenti dopo un mese.
Figura 2 Variazione del numero di giorni di emicrania al mese nello studio 2a]

a Sono indicati la media dei minimi quadrati e gli intervalli di confidenza al 95%.
Il valore p della differenza nella media dei minimi quadrati tra erenumab e il placebo come media nei mesi 4, 5 e 6 (variabile di outcome primaria) era <0.001.
Tabella 2 Variazioni dell'efficacia e delle variabili di outcome riferite dal paziente rispetto all'inizio dello studio (basale) alle settimane 13–24 nello Studio 2
Aimovig 70 mg (n = 312) | Aimovig 140 mg (n = 318) | Placebo (n = 316) | Differenza di trattamento/ | valore pa | |
|---|---|---|---|---|---|
Pazienti responder con ≥50% MMDc Percentuale (%) | 43.3 | 50.0 | 26.6 | ORd 70 mg: 2.13 140 mg: 2.81 | <0.001 |
Numero di giorni con medicamenti acuti specifici per emicrania al mesee Variazione mediab (IC 95%) | -1.13 (‑1.34; ‑0.92) | ‑1.61 (‑1.83; ‑1.40) | ‑0.20 | DT 70 mg: ‑0.94 140 mg: ‑1.42 | <0.001 |
DT = differenza di trattamento; IC = intervallo di confidenza; MMD = giorni di emicrania al mese; OR = odds ratio a Tutti i valori p sono riportati come valori p non regolati, che sono statisticamente significativi dopo il raccordo per confronti multipli. b La variazione dei minimi quadrati al basale nei mesi 4–6, la differenza di trattamento e il valore p si basano su un modello di effetti di miscela lineare, tenendo conto di gruppo di trattamento, valore al basale, fattori di stratificazione (regione [Nord America vs. resto del mondo] e precedente uso di medicamenti profilattici [nessun uso, solo uso precedente, uso concomitante]), visita programmata e interazione tra gruppo di trattamento e visita programmata senza imputazione dei dati mancanti. c I responder sono definiti come pazienti che raggiungono una riduzione ≥50% del numero di giorni di emicrania al mese rispetto all'inizio dello studio (basale). d. L'odd ratio e il valore p per ≥50% di responder nei mesi 4–6 sono basati su un test stratificato di Cochran-Mantel-Haenszel dopo l'imputazione dei dati mancanti come non risposta (non response). e I medicamenti specifici per l'emicrania sono ad esempio i triptani e i derivati dell'ergotamina. | |||||
Farmacocinetica
Assorbimento
Erenumab presenta una cinetica non lineare a causa del legame con il recettore CGRP. La somministrazione sottocutanea di una dose da 70 mg e 140 mg in volontari sani ha prodotto una Cmax media ([deviazione standard, DS]) di 6.1 [2.1] e 15.8 [4.8] μg/ml e un'AUClast media [DS] di 159 [58] e 505 [139] giorni*μg/ml.
I livelli sierici di valle sono aumentati meno del doppio ogni 4 settimane dopo somministrazione sottocutanea di dosi da 70 mg e 140 mg (Cmin [DS] 5.7 [3.1] e 6.2 [2.9] μg/ml con dosaggio di 70 mg e Cmin [DS] 12.8 [6.5] e 14.9 [6.5] μg/ml con dosaggio di 140 mg nei pazienti con emicrania episodica e cronica, rispettivamente) e si sono avvicinati allo stato di equilibrio dinamico dopo 12 settimane di somministrazione.
Dopo la somministrazione di una singola dose per via sottocutanea di 70 mg o 140 mg di Aimovig in adulti sani, il tempo mediano di concentrazione del picco sierico è di circa 6 giorni. La biodisponibilità assoluta stimata dall'analisi farmacocinetica di popolazione è stata dell'82%.
Distribuzione
Dopo la somministrazione di una singola dose per via endovenosa di 140 mg, il volume medio di distribuzione (DS) è stato stimato pari a 3.86 (0.77) litri durante la fase terminale (Vz).
Metabolismo
L'emivita effettiva di Aimovig stimata dall'analisi farmacocinetica di popolazione è di 28 giorni.
Eliminazione
Per Aimovig sono state rilevate due fasi di eliminazione. A basse concentrazioni, l'eliminazione di Aimovig avviene principalmente attraverso il legame saturabile con la molecola target (CGRP-R), a concentrazioni più elevate in gran parte attraverso una via di eliminazione proteolitica non specifica, non saturabile.
Linearità/non linearità
Dopo la somministrazione sottocutanea di dosi mensili da 70 e 140 mg, la farmacocinetica di Aimovig nell'intervallo di dosaggio è lineare con un aumento proporzionale alla dose della concentrazione sierica.
Cinetica di gruppi di pazienti speciali
L'analisi farmacocinetica di popolazione ha rivelato che la farmacocinetica di erenumab non era influenzata dall'età, dal sesso, dall'etnia, dal sottotipo di emicrania (emicrania episodica o cronica) o dalla velocità di filtrazione glomerulare stimata (eGFR) in nessuna delle popolazioni di pazienti approvate. I pazienti con compromissione renale grave (eGFR <30 ml/min/1.73 m2) non sono stati studiati.
Dati preclinici
Sulla base di studi convenzionali di sicurezza farmacologica, tossicità a dosi multiple e tossicità per la riproduzione e lo sviluppo, i dati preclinici non rivelano rischi particolari per l'uomo.
Mutagenicità
Il potenziale di mutagenicità di Aimovig non è stato studiato. Tuttavia, è improbabile che gli anticorpi monoclonali provochino alterazioni di DNA o cromosomi.
Cancerogenicità
Non sono stati condotti studi di cancerogenicità con Aimovig. Aimovig non è farmacologicamente attivo nei roditori ma presenta un'attività biologica nelle scimmie cynomolgus. Tuttavia, questa specie non è un modello adatto per studiare il rischio di sviluppo di tumori.
Tossicità per la riproduzione
In uno studio di riproduzione su scimmie cynomolgus (ePPND, studio avanzato di sviluppo pre- e post-natale), non sono stati rilevati effetti sulla gestazione e sullo sviluppo embrio-fetale o postnatale (fino a 6 mesi di età), se erenumab viene assunto durante la gravidanza a dosi 17 volte più elevate di quelle ottenute in pazienti trattati con 140 mg una volta al mese (in base all'AUC sierica). Sono state osservate concentrazioni sieriche di erenumab misurabili nelle scimmie neonate alla nascita, a conferma del fatto che erenumab, come altri anticorpi IgG, attraversa la barriera placentare.
Altri dati (tossicità locale, fototossicità, immunotossicità)
Negli studi sulla tossicità cronica nelle scimmie adulte che hanno ricevuto una dose sottocutanea fino a 150 mg/kg di Aimovig due volte a settimana per un massimo di 6 mesi, non sono stati osservati effetti indesiderati a esposizioni sistemiche fino a 123 volte superiori rispetto a quando la dose clinica di 140 mg veniva somministrata ogni 4 settimane (in base all'AUC sierica). Inoltre, in questi studi non sono stati riscontrati effetti indesiderati sui marcatori surrogati per la fertilità (alterazioni patologico-anatomiche o istopatologiche negli organi riproduttivi).
Altre indicazioni
Incompatibilità
In assenza di studi di compatibilità, Aimovig non deve essere somministrato in combinazione con altri medicamenti.
Stabilità
Il medicamento non deve essere utilizzato oltre la data indicata con «EXP» sul contenitore. La siringa preriempita e la penna preriempita sono esclusivamente monouso.
Indicazioni particolari concernenti l'immagazzinamento
Conservare in frigorifero (2–8 °C).
Conservare la siringa preriempita o la penna preriempita nella confezione e nella scatola originale per proteggere il contenuto dalla luce.
Conservare fuori dalla portata dei bambini.
Una volta estratto dal frigorifero, Aimovig può essere conservato a temperatura ambiente (15–25 °C) e deve essere utilizzato entro 14 giorni. Una volta raggiunta la temperatura ambiente, Aimovig non deve essere rimesso in frigorifero.
Indicazioni per la manipolazione
Prima dell'uso, il liquido deve essere controllato. Non iniettare se il liquido è torbido o inequivocabilmente giallo o se contiene fiocchi o particelle.
Lasciare Aimovig a temperatura ambiente per almeno 30 minuti prima della somministrazione sottocutanea.
Non agitare il preparato.
Numero dell'omologazione
66620, 66748 (Swissmedic)
Titolare dell’omologazione
Novartis Pharma Schweiz AG. Risch; domicilio: 6343 Rotkreuz
Stato dell'informazione
Agosto 2020
Composition
Aimovig® 70 mg/ml, 140 mg/ml
Principes actifs
Erenumabum (fabriqué par génie génétique en utilisant des cellules ovariennes de hamster chinois).
Excipients
Saccharum, Polysorbatum 80, Natrii hydroxidum corresp. à 0.2 mg/ml de sodium, Acidum aceticum glaciale, Aqua ad iniectabilia.
Forme pharmaceutique et quantité de principe actif par unité
Solution injectable en seringue préremplie ou en stylo prérempli pour injection sous-cutanée:
1 seringue préremplie ou 1 stylo prérempli de 1 ml contient 70 mg d'érénumab dans 1 ml (70 mg/ml) de solution injectable
1 seringue préremplie ou 1 stylo prérempli de 1 ml contient 140 mg d'érénumab dans 1 ml (140 mg/ml) de solution injectable
Indications/Possibilités d’emploi
Traitement prophylactique de la migraine chez l'adulte, pour autant qu'il soit indiqué.
Posologie/Mode d’emploi
L'indication pour le traitement doit être posée par un médecin expérimenté dans le domaine du traitement de la migraine et celui-ci doit accompagner le patient dans le cadre de la poursuite du traitement. En cas de réponse thérapeutique insuffisante, au bout de 12 mois au plus tard, il convient de procéder à une réévaluation de la poursuite du traitement.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Une dose d'Aimovig de 70 mg sous forme d'injection sous-cutanée une fois par mois est recommandée.
Chez les patients montrant une efficacité insuffisante avec cette posologie, cette dernière peut être augmentée à 140 mg une fois par mois tant qu'une efficacité supérieure est ainsi démontrée.
Patients présentant des troubles de la fonction hépatique
Aucune étude clinique n'a été réalisée auprès de patients présentant des troubles de la fonction hépatique. Aucun ajustement de la posologie n'est recommandé chez les patients atteints d'insuffisance hépatique, car l'érénumab, une immunoglobuline G humaine, n'est pas métabolisé par les enzymes du cytochrome P450 et sa clairance n'est pas effectuée majoritairement par le foie.
Patients présentant des troubles de la fonction rénale
Aucun ajustement de la posologie n'est nécessaire chez les patients présentant un dysfonctionnement rénal léger à modéré. L'analyse pharmacocinétique de la population de données intégrées provenant des études cliniques avec Aimovig n'a montré aucune différence entre les patients présentant un dysfonctionnement léger ou modéré et ceux ayant une fonction rénale normale en ce qui concerne la pharmacocinétique de l'érénumab. Les patients présentant un dysfonctionnement rénal sévère (DFGe <30 ml/min/1.73 m2) n'ont pas été étudiés.
Patients âgés
Le nombre de patients de plus de 65 ans ayant été inclus dans les études cliniques n'est pas suffisant pour pouvoir affirmer si de tels patients réagissent autrement que des patients plus jeunes. Un ajustement de la posologie n'est pas nécessaire, l'âge du patient n'ayant pas d'influence sur la pharmacocinétique d'érénumab.
Enfants et adolescents
La sécurité et l'efficacité d'Aimovig chez les enfants et les adolescents n'ont pas été étudiées. Aimovig ne doit donc pas être utilisé dans ce groupe d'âge.
Prise retardée
Au cas où une dose prévue a été oubliée, elle doit être administrée le plus rapidement possible et les injections suivantes doivent être planifiées mensuellement à partir de la date de la dernière injection.
Mode d'administration
Aimovig est destiné à l'auto-administration par le patient dans la paroi abdominale ou dans la cuisse. Il faut alterner les zones d'injection et l'injection ne doit pas être effectuée dans des zones où la peau est sensible, blessée, rougie, ou durcie. Les injections peuvent également être administrées dans le bras par une autre personne.
La personne administrant le médicament doit avoir été formée à la technique d'administration par un spécialiste. De plus amples instructions concernant le mode d'emploi figurent dans l'information destinée aux patients (notice d'emballage).
Contre-indications
Hypersensibilité au principe actif érénumab ou à l'un des excipients selon la composition.
Mises en garde et précautions
Réactions d'hypersensibilité
Des réactions d'hypersensibilité sévères, y compris un rash, un angioedème et des réactions anaphylactoïdes, ont été signalées avec Aimovig depuis sa commercialisation. Ces réactions peuvent survenir en l'espace de quelques minutes, certaines peuvent toutefois également apparaître plus d'une semaine après le traitement. Si une réaction d'hypersensibilité grave ou sévère survient, l'utilisation d'Aimovig doit immédiatement être terminée et un traitement approprié doit être initié.
Constipation accompagnée de complications graves
Aimovig peut provoquer des constipations accompagnées de graves complications (voir «Effets indésirables»).
Les patients qui prennent Aimovig doivent donc faire l'objet d'une surveillance permettant de déceler les signes d'une constipation grave et recevoir un traitement clinique approprié.
La prise concomitante de médicaments associés à une motilité gastro-intestinale réduite peut accroître le risque de constipation grave et de complications potentielles.
Patients souffrant de maladies cardiovasculaires graves
Les patients souffrant de certaines maladies cardiovasculaires ont été exclus de la participation aux études cliniques. Il n'existe pas de données sur la sécurité pour ces patients.
Patients sensibles au latex
Les capuchons amovibles de la seringue préremplie et du stylo prérempli Aimovig contiennent du latex de caoutchouc naturel sec qui peut provoquer des réactions allergiques chez les personnes sensibles au latex.
Ce médicament contient moins de 1 mmol de sodium (23 mg) par seringue préremplie ou par stylo prérempli (70 mg/ml, 140 mg/ml), c'est-à-dire qu'il est pratiquement «sans sodium».
Interactions
Lors d'une étude en ouvert portant sur les interactions médicamenteuses pharmacocinétiques entre Aimovig et un contraceptif oral d'association chez des sujets sains de sexe féminin, l'érénumab (140 mg par voie sous-cutanée [SC], dose unique) n'a eu aucune influence sur la pharmacocinétique d'un contraceptif oral d'association contenant de l'éthinylestradiol et du norgestimate.
Lors d'une étude randomisée, en double aveugle, contrôlée contre placebo chez des sujets sains, l'administration concomitante d'érénumab (140 mg par voie intraveineuse [IV], dose unique) et de sumatriptan par comparaison avec l'administration de sumatriptan seul n'a eu aucune influence sur la pression artérielle au repos. Aimovig n'a pas eu d'influence sur la pharmacocinétique du sumatriptan.
L'érénumab n'est pas métabolisé par les enzymes du cytochrome P450. Des interactions avec des médicaments concomitants qui sont des substrats, des inducteurs ou des inhibiteurs des enzymes du cytochrome P450 sont donc improbables.
Grossesse/Allaitement
Grossesse
Il n'existe pas de données suffisantes concernant l'emploi d'Aimovig chez la femme enceinte. Les expérimentations animales n'ont révélé aucun effet nuisible direct ou indirect en ce qui concerne la toxicité sur la reproduction (voir «Données précliniques»).
Les expérimentations animales ne permettent pas toujours de prévoir la réaction chez l'être humain, on ignore donc si Aimovig est nocif pour le fœtus lorsqu'il est administré à une femme enceinte. Aimovig ne doit pas être utilisé pendant une grossesse, sauf en cas de nécessité absolue.
Allaitement
On ignore si Aimovig passe dans le lait maternel humain. Étant donné que de nombreux médicaments, entre autres les anticorps, sont éliminés dans le lait maternel, un risque pour le nouveau-né ou pour l'enfant en bas âge ne peut pas être exclu. La décision d'arrêter l'allaitement ou la prise d'Aimovig, doit donc être prise en mettant en balance le bénéfice possible d'Aimovig pour la mère et celui de l'allaitement pour le nourrisson.
Fertilité
Il n'existe aucune donnée concernant l'effet d'Aimovig sur la fertilité chez l'être humain. Les expérimentations animales n'ont révélé aucun signe d'effets nuisibles concernant la fertilité (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machines
Aucune étude correspondante n'a été effectuée. Sur la base des données disponibles, on s'attend cependant à ce qu'Aimovig n'ait aucune influence sur l'aptitude à la conduite ou l'utilisation de machines.
Effets indésirables
Pour l'évaluation de la sécurité d'Aimovig par comparaison à un placebo jusqu'à 12 semaines après le début du traitement, les données de deux études cliniques de phase 3 et de deux études cliniques de phase 2 sur la migraine ont été combinées.
2 656 patients au total ont participé aux études contrôlées contre placebo. 1 613 patients ont reçu Aimovig et 1 043 patients ont reçu un placebo. Parmi ces premiers, 893 patients ont reçu une dose de 70 mg d'Aimovig et 507 patients ont reçu une dose de 140 mg d'Aimovig.
Le collectif total pour l'évaluation de la sécurité, y compris les patients se trouvant dans la phase d'extension en ouvert encore en cours avec Aimovig, était supérieur à 2 500 patients (>2 300 patients-années) ayant reçu au moins une dose d'Aimovig: plus de 2 000 patients ont été traités pendant au moins 6 mois et plus de 1 200 patients ont été traités pendant au moins 12 mois.
Les fréquences sont définies comme suit:
«très fréquents» (≥1/10), «fréquents» (≥1/100 à <1/10), «occasionnels» (≥1/1000 à <1/100), «rares» (≥1/10 000 à <1/1000), «très rares» (<1/10 000).
Affections du système immunitaire
Fréquence inconnue: réactions d'hypersensibilité, y compris rash, angioedème et réactions anaphylactoïdes (voir «Mises en garde et précautions»).
Affections gastro-intestinales
Fréquents: constipation (voir «Mises en garde et précautions» et «Description de certains effets indésirables»).
Affections de la peau et du tissu sous-cutané
Fréquents: démangeaisons.
Affections musculo-squelettiques et du tissu conjonctif
Fréquents: crampes musculaires.
Troubles généraux et anomalies au site d'administration
Fréquents: réactions au site d'injection (douleurs, érythème ou démangeaisons).
Effets indésirables après commercialisation
Affections du système immunitaire
Fréquence inconnue: réactions d'hypersensibilité, y compris rash, angioedème et réactions anaphylactoïdes (voir «Mises en garde et précautions»).
Affections gastro-intestinales
Fréquence inconnue: des cas de constipation accompagnée de graves complications ont été rapportés (voir «Mise en garde et précautions» et «Description de certains effets indésirables».
Description de certains effets indésirables
Immunogénicité
Lors des 4 études cliniques (20120178, 20120295, 20120296 et 20120297), l'incidence du développement d'anticorps contre l'érénumab pendant la phase de traitement en double aveugle a été de 6.3% (56/884) chez des patients sous traitement par 70 mg d'érénumab (dont 3 pour lesquels une activité neutralisante in vitro a été démontrée) et de 2.6% (13/504) chez des patients sous traitement par 140 mg d'érénumab (pour lesquels aucune activité neutralisante in vitro n'a été démontrée). Les taux résiduels moyens d'érénumab à la 12e semaine chez les patients positifs aux anticorps anti-érénumab étaient de 40% inférieurs à ceux des patients négatifs aux anticorps. Le développement d'anticorps anti-érénumab n'a pas eu d'incidence sur l'efficacité ou la sécurité de l'érénumab.
L'incidence des anticorps anti-médicaments (ADA) dépend fortement de la sensibilité et de la spécificité du test. En outre, l'incidence de la positivité de l'anticorps observée (y compris les anticorps neutralisants) dans un test peut être influencée par plusieurs facteurs, comme par exemple par la méthodologie du test, la manipulation des échantillons, le moment du prélèvement des échantillons, les médicaments concomitants et la maladie de base. Par conséquent, la comparaison de l'incidence des anticorps anti-érénumab avec l'incidence des anticorps contre d'autres médicaments peut être fallacieuse.
Constipation
La constipation a été l'un des effets secondaires les plus fréquents observés dans les études cliniques rapportées au cours de la phase de 12 semaines contrôlée contre placebo (jusqu'à 3% des cas).
Tous les cas étaient de degré léger ou moyen. Après la commercialisation, des cas de constipation accompagnée de graves complications ont en outre été rapportés. Certains cas ont nécessité une hospitalisation, y compris avec intervention chirurgicale. Pour la plupart des patients, la constipation est survenue après la première dose d'Aimovig. Cependant, des cas de constipation ont aussi été rapportés plus tard au cours du traitement. Le traitement par Aimovig a été interrompu dans la plupart des cas de grave constipation rapportés. Un grand nombre de ces cas concernaient des patients avec des antécédents de constipation ou qui utilisaient simultanément des médicaments associés à une motilité gastro-intestinale diminuée (voir «Mise en garde et précautions»).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
Il n'existe aucune expérience de surdosage d'Aimovig lors des études cliniques. Lors des études cliniques, des doses allant jusqu'à 280 mg ont été administrées par voie sous-cutanée sans qu'une toxicité limitant la dose n'ait été constatée.
En cas de surdosage, la personne concernée doit être traitée de façon symptomatique et des mesures d'aides doivent être prises si besoin.
Propriétés/Effets
Code ATC
N02CD01
Mécanisme d'action
Pharmacodynamique
L'érénumab est un anticorps monoclonal antagoniste humain dirigé contre le récepteur du peptide lié au gène de la calcitonine (récepteur CGRP [calcitonin gene-related peptide]) sans effet pharmacologique important sur les récepteurs de l'adrénomédulline, de la calcitonine et de l'amyline et sans effet agoniste sur le récepteur CGRP.
Le CGRP est un neuropeptide qui module la transmission du signal nociceptif et un vasodilatateur qui a été associé à la physiopathologie de la migraine.
Le récepteur CGRP se situe sur de sites qui sont pertinents pour la physiopathologie de la migraine. L'érénumab entre en compétition de manière puissante et spécifique avec le CGRP en termes de liaison au récepteur CGRP et inhibe ainsi la fonction du CGRP au récepteur CGRP.
Lors d'une étude randomisée en double aveugle et contrôlée contre placebo (20140254) visant à évaluer l'efficacité d'Aimovig (140 mg par voie intraveineuse, dose unique) chez des patients atteints d'angine de poitrine stable, Aimovig n'a pas conduit à une diminution de la durée de l'effort lors du test du tapis roulant par comparaison au placebo et n'a pas entraîné d'aggravation de l'ischémie myocardique.
Efficacité clinique
Migraine chronique
Étude 1 (Étude 20120295)
Aimovig a été évalué dans le cadre d'une étude randomisée, multicentrique, d'une durée de 12 semaines, contrôlée contre placebo, en double aveugle sur la prophylaxie de la migraine chronique. En tout, 667 patients souffrant de migraine avec ou sans aura dans leurs antécédents (≥15 jours de céphalées par mois, avec ≥8 jours de migraine par mois) ont été randomisés, pour recevoir toutes les 4 semaines durant 12 semaines un placebo (n = 286), 70 mg d'Aimovig (n = 191) ou 140 mg d'Aimovig (n = 190) par injection sous-cutanée.
Au début de l'étude, la fréquence moyenne des migraines était d'environ 18 jours de migraine par mois et était similaire dans tous les groupes de traitement. Au cours de l'étude, les patients avaient le droit d'utiliser des traitements aigus contre les céphalées, comme par exemple des triptans, des dérivés de l'ergotamine et des AINS.
L'âge médian des patients était de 43 ans (tranche d'âge: 18–66 ans), 83% étaient de sexe féminin et 94% étaient blancs. Les patients ayant souffert d'un infarctus du myocarde, d'un accident vasculaire cérébral, d'accidents ischémiques transitoires, d'une angine de poitrine instable, subi un pontage coronarien ou autres procédures de revascularisation au cours des 12 derniers mois précédant la sélection ont été exclus de l'étude 1.
La variable de résultat principale était la modification des jours de migraine par mois au mois 3 par rapport au début de l'étude. L'obtention d'une diminution des jours de migraine par mois de 50 à 100% (répondeur à ≥50%) par rapport au début de l'étude et la modification du nombre de jours par mois avec recours à une médication aiguë spécifique de la migraine par rapport au début de l'étude comptaient parmi les variables de résultat secondaires.
Illustration 1 Modification du nombre de jours de migraine par mois dans l'étude 1

Sont indiqués les valeurs moyennes des moindres carrés et les intervalle de confiance à 95%.
La valeur p pour la différence des moyennes des moindre carrés entre l'érénumab et le placebo lors de l'évaluation au mois 3 (variable de résultat principale) était <0.001.
Tableau 1 Modifications de l'efficacité et des variables de résultat rapportées par les patients eux-mêmes à la semaine 12 par rapport au début de l'étude (référence) dans l'étude 1
Aimovig 70 mg (n = 188) | Aimovig 140 mg (n = 187) | Placebo (n = 281) | Différence de traitement/ Odds ratioc (IC à 95%) | valeur pa | |
|---|---|---|---|---|---|
répondeurs pour cent (%) | 39.9 | 41.2 | 23.5 | ORd 70 mg: 2.18 (1.46; 3.27) 140 mg: 2.34 (1.56; 3.51) | <0.001 |
Nombre de jours avec recours à une médication aiguë spécifique de la migraine (par mois)e Modification moyenneb (IC à 95%) | ‑3.45 (‑4.02, ‑2.87) | ‑4.13 (‑4.70; ‑3.56) | ‑1.58 (‑2.05; ‑1.11) | DT 70 mg: ‑1.86 (‑2.60, ‑1.13) 140 mg: ‑2.55 (‑3.28; ‑1.82) | <0.001 |
DT = différence de traitement, IC = intervalle de confiance; JMM = jours de migraine par mois, OR = odds ratio a. Toutes les valeurs p sont indiquées en tant que valeurs p non ajustées qui, après ajustement en vue de la comparaison multiple, sont statistiquement significatives. b. La modification des moyennes des moindres carrés par rapport au début de l'étude au mois 3, la différence de traitement et la valeur p reposent sur un modèle linéaire à effets mixtes prenant en compte le groupe de traitements, la valeur par mois au début de l'étude, les facteurs de stratification -(région [Amérique du Nord vs Europe] et l'utilisation excessive de médicaments [disponible vs non disponible]), la visite planifiée et l'interaction entre le groupe de traitement et la visite planifiée sans imputation en cas de données manquantes. c. Les répondeurs sont définis comme les patients qui parviennent à une diminution ≥50% du nombre de jours de migraine par mois par rapport au début de l'étude (référence). d. L'odds ratio et la valeur p pour les répondeurs ≥50% au mois 3 reposent sur un test de Cochran-Mantel-Haenszel stratifié après imputation des données manquantes comme non-réponse (Non-response). e. Des médicaments spécifiques de la migraine sont par exemple les triptans et les dérivés de l'ergotamine. | |||||
Migraine épisodique
Étude 2 (Étude 20120296, STRIVE)
L'étude 2 est une étude randomisée, multicentrique, d'une durée de 24 semaines, contrôlée contre placebo et en double aveugle visant à évaluer Aimovig comme traitement prophylactique de la migraine épisodique. En tout, 955 patients atteints d'une migraine d'une durée supérieure ou égale à 12 mois, avec ou sans aura et 4 à 14 jours de migraine par mois dans les antécédents médicaux, ont été randomisés, afin d'obtenir toutes les 4 semaines durant 24 semaines 70 mg d'Aimovig (n = 317), 140 mg d'Aimovig (n = 319) ou un placebo (n = 319) par injection sous-cutanée.
Au début de l'étude, la fréquence moyenne des migraines était d'environ 8 jours de migraine par mois et était similaire dans tous les groupes de traitement. Au cours de l'étude, les patients avaient le droit d'utiliser des traitements aigus contre les céphalées, comme par exemple des triptans, des dérivés de l'ergotamine et des AINS.
L'âge médian des patients était de 42 ans (18–65 ans), 85% étaient de sexe féminin et 89% étaient blancs. Les patients utilisant des médicaments de manière excessive, les patients ayant souffert d'un infarctus du myocarde, d'un accident vasculaire cérébral, d'accidents ischémiques transitoires, d'une angine de poitrine instable, subi un pontage coronarien ou autres procédures de revascularisation au cours des 12 derniers mois précédant la sélection ont été exclus de la participation à l'étude 2.
Le critère d'évaluation principal était la modification des jours de migraine durant les mois 4 à 6 par rapport au début de l'étude. Les critères d'évaluation secondaires étaient entre autres l'obtention d'une diminution du nombre moyen des jours de migraine par mois de 50 à 100% (répondeur à ≥50%) par rapport au début de l'étude et la modification du nombre moyen de jours par mois avec recours à une médication aiguë spécifique de la migraine par rapport au début de l'étude.
Des différences par rapport au placebo ont déjà été observées au bout d'un mois.
Illustration 2 Modification du nombre de jours de migraine par mois dans l'étude 2a

a Les indications sont les moyennes des moindres carrés et les intervalles de confiance à 95%.
La valeur p pour la différence des moyennes des moindres carrés entre l'érénumab et le placebo comme valeur moyenne des mois 4, 5 et 6 (variable de résultat principale) était <0.001
Tableau 2 Modifications de l'efficacité et des variables de résultat rapportées par les patients eux-mêmes par rapport au début de l'étude (référence) au cours des semaines 13 à 24 dans l'étude 2
Aimovig 70 mg (n = 312) | Aimovig 140 mg (n = 318) | Placebo (n = 316) | Différence de traitement/Odds ratio (IC à 95%) | valeur pa | |
|---|---|---|---|---|---|
répondeurs JMM ≥50% c pour cent (%) | 43.3 | 50.0 | 26.6 | ORd 70 mg: 2.13 140 mg: 2.81 | <0.001 |
Nombre de jours avec recours à une médication aiguë spécifique de la migraine par moise Modification moyenneb | ‑1.13 (‑1.34; ‑0.92) | ‑1.61 (‑1.83; ‑1.40) | ‑0.20 | DT 70 mg:‑0.94 (‑1.23; ‑0.64) 140 mg: ‑1.42 | <0.001 |
DT = différence de traitement, IC = intervalle de confiance; JMM = jours de migraine par mois, OR = odds ratio a Toutes les valeurs p sont données en tant que valeurs p non ajustées qui, après ajustement en vue de la comparaison multiple, sont statistiquement significatives. b La modification des moyennes des moindres carrés par rapport au début de l'étude au cours des mois 4 à 6, la différence de traitement et la valeur p reposent sur un modèle linéaire à effets mixtes prenant en compte le groupe de traitements, la valeur au début de l'étude, les facteurs de stratification (région [Amérique du Nord vs le reste du monde] et l'utilisation préalable de médicaments prophylactiques [pas d'utilisation, seulement une utilisation préalable, une utilisation concomitante]), la visite planifiée et l'interaction entre le groupe de traitement et la visite planifiée sans imputation en cas de données manquantes. c Les répondeurs sont définis comme des patients qui parviennent à une diminution ≥50% du nombre de jours de migraine par mois par rapport au début de l'étude (référence). d L'odds ratio et la valeur p pour des répondeurs ≥50%au cours des mois 4 à 6 reposent sur un test de Cochran-Mantel-Haenszel stratifié après imputation des données manquantes comme non-réponse (Non-response). e Des médications spécifiques de la migraine sont par exemple les triptans et les dérivés de l'ergotamine. | |||||
Pharmacocinétique
Absorption
L'érénumab présente une cinétique non linéaire en raison de la liaison au récepteur CGRP. L'administration sous-cutanée d'une dose de 70 mg et 140 mg à des sujets sains a conduit à une valeur moyenne de Cmax (écart-type [standard deviation, SD]) de 6.1 [2.1] et 15.8 [4.8] µg/ml et à une valeur moyenne de l'AUClast [SD] de 159 [58] et de 505 [139] jour*µg/ml.
Les taux résiduels sériques après l'administration sous-cutanée de doses de 70 mg et 140 mg toutes les 4 semaines ont augmenté de moins du double (Cmin [SD] 5.7 [3.1] et 6.2 [2.9] µg/ml avec une posologie de 70 mg et Cmin [SD] 12.8 [6.5] et 14.9 [6.5] µg/ml avec une posologie de 140 mg chez des patients atteints de migraine épisodique ou chronique) et se sont rapprochés de l'état stationnaire après une administration pendant 12 semaines.
Suite à l'administration sous-cutanée d'une dose unique de 70 mg ou de 140 mg d'Aimovig à des adultes sains, la durée médiane jusqu'au pic de concentration sérique a été d'environ 6 jours. La biodisponibilité absolue estimée sur la base de l'analyse pharmacocinétique de population était de 82%.
Distribution
Après administration intraveineuse d'une dose unique de 140 mg, le volume moyen de distribution (SD) pendant la phase terminale (Vz) a été estimé à 3.86 (0.77) litres.
Métabolisme
La demi-vie effective d'Aimovig estimée sur la base de l'analyse pharmacocinétique de population est de 28 jours.
Élimination
Deux phases d'élimination ont été observées pour Aimovig. À faibles concentrations, l'élimination d'Aimovig a lieu principalement par liaison saturable à la molécule cible (récepteur CGRP), à des concentrations plus élevées en grande partie par une voie d'élimination protéolytique non spécifique et non saturable.
Linéarité/non-linéarité
Après administration sous-cutanée mensuelle de doses de 70 et 140 mg, la pharmacocinétique d'Aimovig dans l'intervalle posologique est linéaire avec une augmentation de la concentration sérique proportionnelle à la dose.
Cinétique pour certains groupes de patients
L'analyse pharmacocinétique (PK) de population a permis de conclure que la pharmacocinétique de l'érénumab n'a été influencée dans aucun des groupes de patients autorisés en fonction de l'âge, du sexe, de l'ethnie, du sous-type de migraine (migraine épisodique ou chronique) ou du débit de filtration glomérulaire estimé (DFGe). Les patients présentant un dysfonctionnement rénal sévère (DFGe <30 ml/min/1.73 m2) n'ont pas été étudiés.
Données précliniques
Sur la base d'études conventionnelles de pharmacologie de sécurité, de la toxicité de dosages multiples et de la toxicité sur la reproduction et le développement, les données précliniques ne permettent d'identifier aucun risque particulier pour l'être humain.
Mutagénicité
Le potentiel de mutagénicité d'Aimovig n'a pas été étudié. Cependant, il est peu probable que des anticorps monoclonaux modifient l'ADN ou les chromosomes.
Carcinogénicité
Aucune étude sur la cancérogénicité n'a été effectuée avec Aimovig. Aimovig n'est pas actif pharmacologiquement chez les rongeurs, mais présente une activité biologique chez les singes cynomolgus; cette espèce animale n'est cependant pas un modèle approprié pour l'étude du risque de formation de tumeur.
Toxicité sur la reproduction
Lors d'une étude sur la reproduction réalisée chez des singes cynomolgus (étude sur le développement pré- et postnatal renforcé (ePPND, enhanced pre- and postnatal development), aucune incidence sur la gestation et sur le développement embryo-fœtal ou postnatal (jusqu'à l'âge de 6 mois) n'a été constatée lorsque l'érénumab a été administré à des animaux pendant la gestation à des doses qui ont entraîné une exposition 17 fois plus élevée que celle obtenue chez des patients sous traitement avec 140 mg une fois par mois (en se basant sur la valeur de l'AUC sérique). Chez les singes nouveau-nés, l'érénumab a été détecté dans le sérum à la naissance, ce qui confirme que l'érénumab, comme d'autres anticorps IgG, traverse la barrière placentaire.
Autres données (toxicité locale, phototoxicité, immunotoxicité)
Lors d'études de toxicité chronique chez des singes sexuellement matures auxquels une dose allant jusqu'à 150 mg/kg d'Aimovig a été administrée par voie sous-cutanée deux fois par semaine sur une durée maximale de 6 mois, aucun effet indésirable n'a été constaté pour des expositions systémiques qui étaient jusqu'à 123 fois plus élevées que dans le cas de l'administration de la dose clinique de 140 mg toutes les 4 semaines (en se basant sur la valeur de l'AUC sérique). De plus, aucun effet indésirable sur des marqueurs de substitution pour la fertilité (modifications pathologiques et anatomiques ou histopathologiques au niveau des organes de reproduction) n'a été rapporté lors de ces études.
Remarques particulières
Incompatibilités
Aucune étude de compatibilité n'étant existante, ce médicament ne doit pas être mélangé à d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient. La seringue préremplie ou le stylo prérempli sont destinés à un usage unique.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2–8 °C)
Conserver la seringue préremplie ou le stylo prérempli dans l'emballage d'origine, dans le carton, pour protéger le contenu de la lumière.
Conserver hors de portée des enfants.
Après avoir retiré Aimovig du réfrigérateur, il peut être conservé à température ambiante (15–25 °C) et doit ensuite être utilisé dans un délai de 14 jours. Aimovig ne doit pas être conservé à nouveau dans le réfrigérateur après avoir été mis à température ambiante.
Remarques concernant la manipulation
Le liquide doit être contrôlé avant l'utilisation. Ne pas injecter si le liquide est trouble ou nettement jaune ou s'il contient des flocons ou des particules.
Avant l'administration sous-cutanée, Aimovig doit être placé au moins 30 minutes à température ambiante.
Ne pas agiter la préparation.
Numéro d’autorisation
66620, 66748 (Swissmedic)
Titulaire de l’autorisation
Novartis Pharma Schweiz AG Risch; domicile: 6343 Rotkreuz
Mise à jour de l’information
Août 2020
Відгуки (0)

Безкоштовна консультація досвідченого спеціаліста
Опишіть симптоми або потрібний продукт - ми допоможемо підібрати його дозування або аналог, оформити замовлення з доставкою додому або просто проконсультуємо.
Нас 14 спеціалістів і 0 ботів. Ми завжди будемо з вами на зв'язку і зможемо зв'язатися в будь-який час.
Хіт продажів


HerpoTherm ручка від герпесу
Код продукту: 7798882Herpotherm® - нагрівальна ручка Герпез не тільки непривабливий, але й може бути дуже болючим. Супут..
8214.70 RUB


Extra Cell Matrix C-II TABS для суглобів 120 шт
Код продукту: 5677150Extra Cell Matrix C-II TABS для суглобів 120 шт Extra Cell Matrix C-II TABS for Joints – це високоеф..
13106.98 RUB






Vita Omexanthin капсули 60 шт
Код продукту: 6161945Vita Omexanthin капсули - 60 шт Капсули Vita Omexanthin - це дієтична добавка з комбінацією омега-3 ..
16063.71 RUB




Альгіфор-Л форте таблетки плівка 400 мг 10 шт
Код продукту: 3398902..
3515.40 RUB

АЛЬПІНАМЕД Крем для рук і нігтів Tb 100 мл
Код продукту: 1811321ALPINAMED Крем для рук і нігтів ALPINAMED Hand & Nail Cream — це крем для рук, який був спеціаль..
2633.76 RUB



Зубна щітка Trisa Flexible Head жорстка
Код продукту: 2841175Зубна щітка Trisa Flexible Head жорстка Жорстка зубна щітка Trisa Flexible Head ідеально підходить д..
989.74 RUB





Пастилки Phytopharma Islandica без цукру 40 шт
Код продукту: 2561234Пастилки Phytopharma Islandica без цукру 40 шт Опис товару: Пастилки Phytopharma Islandica без цукру..
1808.99 RUB

