




Enspryng Inj Lös 120 мг/мл Ферцпр 1 мл
Enspryng Inj Lös 120 mg/ml Fertspr 1 ml
-
1332039.02 RUB
Дата доставки:
25.12.2025 - 08.01.2026
При оплате криптовалютой:
Ваша прибыль 133203.90 RUB / 1572.25 USDT

- Наявність: В наявності
- Виробник: ROCHE PHARMA (SCHWEIZ)
- Модель: 7779634
- ATC-код L04AC19
- EAN 7680676170011
Склад:
Наведіть телефон на qr-код

Опис
 Deutsch
Deutsch French
French Italian
Italian▼ Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Sie können dabei helfen, indem Sie Nebenwirkungen melden. Hinweise zur Meldung von Nebenwirkungen, siehe Ende Kapitel «Welche Nebenwirkungen kann Enspryng haben?».
Was ist Enspryng und wann wird es angewendet?
Enspryng enthält den Wirkstoff Satralizumab, der zu einer Gruppe von Arzneimitteln gehört, welche als «monoklonale Antikörper» bezeichnet werden. Monoklonale Antikörper sind eine Art von Protein und dazu ausgelegt, eine bestimmte Zielsubstanz im Körper zu erkennen und daran zu binden.
Enspryng ist zur Behandlung von Neuromyelitis-optica-Spektrum-Erkrankungen (NMOSD) bei Erwachsenen und Jugendlichen bestimmt.
NMOSD sind Autoimmunerkrankungen des Zentralnervensystems (ZNS), von denen hauptsächlich die Sehnerven und das Rückenmark betroffen sind. Die Schädigung der Sehnerven führt zu Schwellungen und Entzündungen, die Schmerzen und einen Verlust des Sehvermögens verursachen. Die Schädigung des Rückenmarks führt zu einer Schwäche in Beinen oder Armen oder zum Verlust der Fähigkeit, die Beine oder Arme zu bewegen, zu Gefühllosigkeit und zu Problemen mit der Blasen- und Darmfunktion.
Ein Rezidiv bzw. eine «Attacke» von NMOSD tritt auf, wenn eine Entzündung im Nervensystem vorliegt. Aufgrund der Entzündung treten bei den Betroffenen neue oder aber bekannte, bereits vormals aufgetretene Symptome auf.
Enspryng blockiert die Wirkung eines bestimmten Proteins, einem Zytokin, das als Interleukin-6 (IL-6) bezeichnet wird und an Entzündungsvorgängen im Körper beteiligt ist. Es wurde gezeigt, dass dieses Arzneimittel das Risiko des Auftretens eines Rezidivs von NMOSD verringert.
Auf Verschreibung des Arztes oder der Ärztin.
Wann darf Enspryng nicht angewendet werden?
Wenn Sie allergisch gegen Satralizumab oder einen der in Rubrik «Was ist in Enspryng enthalten» genannten sonstigen Bestandteile dieses Arzneimittels sind.
Wenn Sie sich nicht sicher sind, sprechen Sie mit Ihrem Arzt bzw. Ihrer Ärztin, Ihrem Apotheker oder Ihrer Apothekerin oder dem medizinischen Fachpersonal, bevor Sie Enspryng anwenden.
Wann ist bei der Anwendung von Enspryng Vorsicht geboten?
Sprechen Sie mit Ihrem Arzt bzw. Ihrer Ärztin, Ihrem Apotheker oder Ihrer Apothekerin oder dem medizinischen Fachpersonal, bevor Sie Enspryng anwenden, wenn einer der folgenden Punkte auf Sie zutrifft (oder wenn Sie nicht sicher sind, ob einer der folgenden Punkte auf Sie zutrifft):
- Bei Ihnen liegt eine Infektion vor. Ihr Arzt bzw. Ihre Ärztin wird warten, bis die Infektion abgeklungen ist, bevor Enspryng bei Ihnen angewendet wird.
- Sie wurden kürzlich geimpft.
Enspryng kann Nebenwirkungen verursachen, über die Sie Ihren Arzt bzw. Ihre Ärztin in Kenntnis setzen müssen. Zu diesen gehören:
Infektionen
- Sprechen Sie mit Ihrem Arzt bzw. Ihrer Ärztin, bevor Sie Enspryng erhalten, wenn Sie glauben, dass bei Ihnen eine Infektion vorliegt. Ihr Arzt bzw. Ihre Ärztin wird warten, bis die Infektion abgeklungen ist, bevor Enspryng bei Ihnen angewendet wird.
- Informieren Sie unverzüglich Ihren Arzt bzw. Ihre Ärztin oder das medizinische Fachpersonal, wenn bei Ihnen während oder nach der Enspryng-Behandlung eines dieser Anzeichen einer Infektion auftritt:
- Fieber oder Schüttelfrost
- anhaltender Husten
- Halsschmerzen oder neue offene Stellen (z.B. Fieberbläschen).
- Informieren Sie Ihren Arzt bzw. Ihrer Ärztin, wenn Sie an Grunderkrankungen leiden oder gelitten haben (z.B. Divertikulitis, Diabetes, interstitielle Lungenerkrankungen, Tuberkulose, Hepatitis B), da diese das Auftreten von Infektionen begünstigen können.
Impfungen
- Informieren Sie Ihren Arzt bzw. Ihre Ärztin, wenn Sie kürzlich geimpft wurden oder vielleicht in naher Zukunft geimpft werden.
- Während der Behandlung mit Enspryng sollten Sie keine Lebendimpfstoffe oder abgeschwächte Lebendimpfstoffe erhalten (z.B. gegen Tuberkulose oder Impfstoffe gegen Gelbfieber).
- Ihr Arzt bzw. Ihre Ärztin wird überprüfen, ob Sie Impfungen benötigen, bevor Sie mit der Behandlung mit Enspryng beginnen.
Überempfindlichkeitsreaktionen
- Wenn es während oder nach der Injektion zu einer Überempfindlichkeitsreaktion kommt, kontaktieren Sie umgehend Ihren Arzt bzw. Ihre Ärztin (siehe auch «Rubrik Welche Nebenwirkungen kann Enspryng haben?»).
- Verabreichen Sie sich die nächste Dosis erst, nachdem Sie Ihren Arzt bzw. Ihre Ärztin informiert haben UND Ihr Arzt bzw. Ihre Ärztin Sie zur Anwendung der nächsten Dosis aufgefordert hat.
Leberenzyme
- Enspryng kann dazu führen, dass sich die Menge einiger Leberenzyme in Ihrem Blut während der Behandlung erhöht. Ihr Arzt bzw. Ihre Ärztin wird Blutuntersuchungen durchführen, um diese Mengen zu überprüfen und festzustellen, wie gut Ihre Leber funktioniert.
- Informieren Sie unverzüglich Ihren Arzt bzw. Ihre Ärztin oder das medizinische Fachpersonal, wenn bei Ihnen während oder nach der Enspryng-Behandlung eines dieser Anzeichen für erhöhte Leberenzyme auftritt:
- Gelbfärbung der Haut und des Augenweisses (Gelbsucht)
- dunkler Urin
- Krankheitsgefühl und Krankheitszustand.
Neutrophile
- Enspryng kann dazu führen, dass sich die Anzahl bestimmter weisser Blutkörperchen (Neutrophile Granulozyten, eine Form spezialisierter Immunzellen) in Ihrem Blut während der Behandlung verringert. Ihr Arzt bzw. Ihre Ärztin wird Blutuntersuchungen durchführen, um die Anzahl der weissen Blutkörperchen zu kontrollieren.
Malignome
- Wenn Sie an Krebs leiden, informieren Sie Ihren Arzt bzw. Ihre Ärztin. Ihr Arzt bzw. Ihre Ärztin wird darüber entscheiden, ob eine Behandlung mit Enspryng gleichwohl in Frage kommt.
Lipidparameter
- Wenn bei Ihnen bekannte kardiovaskuläre (Herz-Kreislauf) Risikofaktoren bestehen, wie z.B. ein erhöhter Blutdruck oder zu hohe Cholesterinwerte, informieren Sie Ihren Arzt bzw. Ihre Ärztin. Diese Faktoren müssen während einer Behandlung mit Enspryng gegebenenfalls kontrolliert werden.
Kinder und Jugendliche
Dieses Arzneimittel darf nicht an Kinder unter 12 Jahren verabreicht werden, da es in dieser Altersgruppe noch nicht untersucht worden ist.
Verkehrstüchtigkeit und Fähigkeit zum Bedienen von Maschinen
Es sind keine Auswirkungen auf die Verkehrstüchtigkeit oder die Fähigkeit zum Bedienen von Maschinen beobachtet worden.
Einnahme von Enspryng zusammen mit anderen Arzneimitteln
Informieren Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, wenn Sie
- an anderen Krankheiten leiden,
- Allergien haben oder
- andere Arzneimittel (auch selbstgekaufte!) einnehmen oder äusserlich anwenden (Externa!).
Darf Enspryng während einer Schwangerschaft oder in der Stillzeit angewendet werden?
Empfängnisverhütung (Frauen)
Frauen müssen während der Behandlung und 5 Monate lang nach der letzten Enspryng-Dosis eine Methode zur Empfängnisverhütung anwenden.
Schwangerschaft und Stillzeit
Wenn Sie schwanger sind oder stillen, oder wenn Sie vermuten, schwanger zu sein, oder beabsichtigen, schwanger zu werden, fragen Sie vor der Einnahme dieses Arzneimittels Ihren Arzt bzw. Ihre Ärztin oder Ihren Apotheker bzw. Ihre Apothekerin um Rat.
Möglicherweise wird Ihnen von Ihrem Arzt bzw. Ihrer Ärztin geraten abzustillen, wenn Sie mit Enspryng behandelt werden sollen. Es ist nicht bekannt, ob Enspryng in die Muttermilch übergeht.
Wie verwenden Sie Enspryng?
Verwenden Sie dieses Arzneimittel immer genau so, wie Ihr Arzt bzw. Ihre Ärztin oder Ihr Apotheker bzw. Ihre Apothekerin es Ihnen verordnet hat. Fragen Sie bei Ihrem Arzt bzw. Ihrer Ärztin oder Ihrem Apotheker bzw. Ihrer Apothekerin nach, wenn Sie sich nicht sicher sind.
Enspryng wird als Spritze (Injektion) unter die Haut (subkutan) gegeben. Zu Beginn kann die Enspryng-Injektion durch Ihren Arzt, bzw. Ihre Ärztin oder das medizinische Fachpersonal gegeben werden. Vielleicht ist Ihr Arzt bzw. Ihre Ärztin der Meinung, dass Sie sich Enspryng selber spritzen (injizieren) können. In diesem Falle werden Sie geschult, wie Sie sich Enspryng selber injizieren können. Sprechen Sie mit Ihrem Arzt bzw. Ihrer Ärztin oder dem medizinischen Fachpersonal, wenn Sie Fragen zur Selbstinjektion haben. Ausführliche «Anwendungshinweise» finden Sie am Ende dieser Packungsbeilage.
Für die ersten drei Injektionen beträgt die empfohlene Dosis 120 mg (der gesamte Inhalt der Fertigspritze) einmal alle zwei Wochen (Aufsättigungsdosen). Die erste Injektion wird unter Aufsicht Ihres Arztes bzw. Ihrer Ärztin oder des medizinischen Fachpersonals gegeben.
Nach Gabe der Aufsättigungsdosen beträgt die empfohlene Dosis 120 mg alle vier Wochen (Erhaltungsdosis). Wenden Sie Enspryng so lange einmal alle vier Wochen an, wie Ihr Arzt bzw. Ihre Ärztin es anordnet.
Die empfohlene Dosis ist für alle Patienten gleich, auch für Jugendliche ab 12 Jahren.
Wenn Sie eine grössere Menge von Enspryng angewendet haben, als Sie sollten
Da Enspryng in einer Fertigspritze gegeben wird, ist es unwahrscheinlich, dass Sie eine zu hohe Menge erhalten. Wenn Sie jedoch besorgt sind, wenden Sie sich an Ihren Arzt bzw. Ihre Ärztin, Ihren Apotheker oder Ihre Apothekerin oder an das medizinische Fachpersonal.
Wenn Sie Enspryng versehentlich häufiger spritzen, als Ihnen von Ihrem Arzt bzw. Ihrer Ärztin gesagt worden ist, sollten Sie sich mit Ihrem Arzt bzw. Ihrer Ärztin in Verbindung setzen. Bringen Sie stets den Umkarton oder die Durchstechflasche mit dem Arzneimittel mit, auch wenn diese bereits leer sind.
Wenn Sie die Anwendung von Enspryng vergessen haben
Wenn Sie Ihre Injektionen von Ihrem Arzt bzw. Ihrer Ärztin oder dem medizinischen Fachpersonal erhalten und einen Termin auslassen, vereinbaren Sie unverzüglich einen neuen Termin.
Wenn Sie sich Enspryng selbst und nicht unter der Aufsicht Ihres Arztes bzw. Ihrer Ärztin oder des medizinischen Fachpersonals spritzen und eine Injektion auslassen, sollten Sie diese so bald wie möglich nachholen. Warten Sie nicht bis zur nächsten geplanten Dosis.
Damit die Behandlung ihre volle Wirksamkeit entfalten kann, ist es sehr wichtig, die Injektionen weiterhin durchzuführen. Nachdem Sie die ausgelassene Dosis verabreicht haben, spritzen Sie sich die nächste Dosis entweder
- nach zwei Wochen (Aufsättigungsdosis) oder
- nach vier Wochen (Erhaltungsdosen).
Fragen Sie bei Ihrem Arzt bzw. Ihrer Ärztin oder bei Ihrem Apotheker bzw. Ihrer Apothekerin oder dem medizinischen Fachpersonal nach, wenn Sie sich nicht sicher sind.
Wenn Sie die Anwendung von Enspryng abbrechen
Beenden Sie die Anwendung von Enspryng nicht kurzfristig, ohne vorher mit Ihrem Arzt bzw. Ihrer Ärztin gesprochen zu haben. Wenn Sie weitere Fragen zur Einnahme dieses Arzneimittels haben, wenden Sie sich an Ihren Arzt bzw. Ihre Ärztin oder an Ihren Apotheker bzw. Ihre Apothekerin oder an das medizinische Fachpersonal.
Die Anwendung und Sicherheit von Enspryng bei Kindern unter 12 Jahren ist bisher nicht geprüft worden.
Ändern Sie nicht von sich aus die verschriebene Dosierung. Wenn Sie glauben, das Arzneimittel wirke zu schwach oder zu stark, so sprechen Sie mit Ihrem Arzt oder Apotheker bzw. mit Ihrer Ärztin oder Apothekerin.
Welche Nebenwirkungen kann Enspryng haben?
Wie alle Arzneimittel kann auch dieses Arzneimittel Nebenwirkungen haben, die aber nicht bei jedem auftreten müssen.
Die folgenden Nebenwirkungen sind bei Anwendung dieses Medikaments berichtet worden:
Injektionsbedingte Reaktionen
- Injektionsbedingte Reaktionen sind die häufigste Nebenwirkung einer Behandlung mit Enspryng (sehr häufig: können mehr als 1 von 10 Personen betreffen). In den meisten Fällen handelt es sich um leichte Reaktionen, mitunter können aber auch schwerwiegende Reaktionen auftreten.
- Informieren Sie unverzüglich Ihren Arzt bzw. Ihre Ärztin oder das medizinische Fachpersonal, falls bei Ihnen bis zu 24 Stunden nach der Injektion Anzeichen oder Symptome einer injektionsbedingten Reaktion auftreten. Es können unter anderem die folgenden Symptome auftreten:
- eine Reaktion an der Injektionsstelle (Rötung, Juckreiz, Schmerzen)
- Kopfschmerzen
- Durchfall
- Hitzewallung (Flushing)
- Hautausschlag oder Hautrötung
- juckende Haut
- Reizung oder Schmerzen im Rachen
- Nesselausschlag
- Kurzatmigkeit
- Schwellung des Rachens
- niedriger Blutdruck (Schwindel und Benommenheit)
- Fieber
- Müdigkeits- oder Schwindelgefühl
- Übelkeit
- schneller Herzschlag.
Sonstige Nebenwirkungen:
Informieren Sie Ihren Arzt bzw. Ihre Ärztin oder das medizinische Fachpersonal, wenn Sie eine der folgenden Nebenwirkungen bemerken:
Sehr häufig (betrifft mehr als einen von 10 Anwendern)
- Kopfschmerzen
- Gelenkschmerzen
- Verringerte Konzentration weisser Blutkörperchen
- Harnwegsinfekte
- Infektion der Atemwege
Häufig (betrifft 1 bis 10 von 100 Anwendern)
- Muskelschmerzen
- Steifigkeit
- Migräne
- Taubheitsgefühl
- Schwellung in den Unterschenkeln, Füssen oder Händen
- Hautausschlag oder Juckreiz
- Allergien oder Heuschnupfen
- Niedrige Blutspiegel an Fibrinogen (ein Protein, das an der Blutgerinnung beteiligt ist)
- Erhöhte Konzentration von Cholesterin und Triglyceriden im Blut
- Erhöhte Konzentration von Leberenzymen (Transaminasen) im Blut
- Gelbliche Haut und Augen (Bilirubin im Blut erhöht)
- Verringerte Konzentration von Blutplättchen im Blut
- Anstieg des Körpergewichts
- Grippe
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Dies gilt insbesondere auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind.
Was ist ferner zu beachten?
Das Arzneimittel darf nur bis zu dem auf der Faltschachtel und auf der Etikette der Fertigspritze mit «EXP» bezeichneten Datum verwendet werden.
Lagerungshinweis
Im Kühlschrank (2-8 °C) lagern.
Nicht schütteln. Nicht einfrieren.
- Enspryng kann ungeöffnet über eine einmalige Dauer von bis zu 8 Tagen bei Raumtemperatur nicht über 30 °C ausserhalb des Kühlschranks in der Umverpackung aufbewahrt werden.
- Nach der Aufbewahrung bei Raumtemperatur sollte das Präparat entweder verwendet oder entsorgt werden.
Den Behälter im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Enspryng ist eine farblose bis hellgelbe Flüssigkeit. Sie dürfen dieses Arzneimittel nicht verwenden, wenn Sie eine Trübung, Verfärbung oder sichtbare Schwebstoffe darin feststellen.
Überprüfen Sie die Fertigspritze und den Nadelschutz auf etwaige Beschädigungen. Bei Vorhandensein von Rissen oder Defekten nicht verwenden.
Nachdem die Schutzkappe entfernt wurde, muss die Injektion innerhalb von 5 Minuten begonnen werden, um zu verhindern, dass das Arzneimittel austrocknet und die Nadel verstopft. Wenn die Fertigspritze nicht innerhalb von 5 Minuten nach dem Abnehmen der Schutzkappe verwendet wird, müssen Sie sie in einem durchstichfesten Entsorgungsbehälter für scharfe und spitze Gegenstände entsorgen und eine neue Fertigspritze verwenden.
Weitere Hinweise
Entsorgen Sie Arzneimittel nicht im Abwasser oder Haushaltsabfall. Fragen Sie Ihren Apotheker bzw. Ihre Apothekerin, wie das Arzneimittel zu entsorgen ist, wenn Sie es nicht mehr verwenden. Sie tragen damit zum Schutz der Umwelt bei.
Weitere Auskünfte erteilt Ihnen Ihr Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Diese Personen verfügen über die ausführliche Fachinformation.
Was ist in Enspryng enthalten?
Injektionslösung in einer Fertigspritze.
Wirkstoffe
Der Wirkstoff ist Satralizumab. Jede Fertigspritze enthält 120 mg Satralizumab in 1 ml.
Hilfsstoffe
Die sonstigen Bestandteile sind L-Histidin, L-Asparaginsäure, L-Argininhydrochlorid, Poloxamer 188, Wasser für Injektionszwecke.
Zulassungsnummer
67617 (Swissmedic).
Wo erhalten Sie Enspryng? Welche Packungen sind erhältlich?
In Apotheken gegen ärztliche Verschreibung, die nur zum einmaligen Bezug berechtigt.
Jede Packung Enspryng enthält 1 Fertigspritze.
Zulassungsinhaberin
Roche Pharma (Schweiz) AG, Basel.
Diese Packungsbeilage wurde im Juli 2020 letztmals durch die Arzneimittelbehörde (Swissmedic) geprüft.
Was Sie wissen müssen, um Ihre Enspryng Fertigspritze sicher zu benutzen
Lesen Sie diese Anwendungshinweise:
|
Der Grund dafür ist, dass sie möglicherweise neue Informationen enthalten.
|
Wichtige Hinweise
Stets beachten:
Stets beachten:
|
Wie soll die Enspryng Fertigspritze aufbewahrt werden?
|
Stets beachten:
|
Erforderliches Zubehör für die Injektion Jeder Umkarton mit Enspryng enthält:
Im Umkarton nicht enthalten:
|
Enspryng Fertigspritze (Abbildung A und Abbildung B) Vor dem Gebrauch:
Abbildung A Nach dem Gebrauch:
Abbildung B Die Spritze verfügt über einen Nadelschutz, der die Nadel nach Abschluss der Injektion automatisch abdeckt. |



Vorbereitungen zur Anwendung von Enspryng
1. Den Umkarton mit der Spritze aus dem Kühlschrank nehmen und auf eine saubere, ebene Arbeitsfläche (z. B. einen Tisch) legen.
2. Das Verfalldatum auf der Rückseite des Umkartons überprüfen (Abbildung C). Nicht verwenden, wenn das auf dem Umkarton angegebene Verfallsdatum überschritten ist.
3. Überprüfen, ob das Siegel an der Vorderseite des Umkartons noch intakt ist (Abbildung C). Nicht verwenden, wenn das Siegel beschädigt ist.
Nicht verwenden, wenn das Verfalldatum abgelaufen oder das Siegel nicht mehr intakt ist. In einem solchen Fall mit Schritt 21 «Enspryng entsorgen» fortfahren und Ihren Arzt bzw. Ihre Ärztin kontaktieren.

Abbildung C
4. Den versiegelten Umkarton öffnen (Abbildung D).

Abbildung D
5. Die Spritze vorsichtig am Zylinder anfassen und aus dem Umkarton nehmen (Abbildung E).
Stets beachten:
- Den Umkarton nicht umdrehen, um die Spritze herauszunehmen.
- Nicht die Aktivierungssicherungen berühren. Die Spritze könnte dadurch beschädigt werden.
- Die Spritze nicht am Kolben und nicht am Nadelschutz festhalten.

Abbildung E
Die Spritze überprüfen
(Abbildung F)
6. Das Verfalldatum (EXP) auf der Spritze überprüfen. Die Spritze nicht verwenden, wenn sie abgelaufen ist.
7. Die Spritze auf Beschädigungen überprüfen. Bei Vorhandensein von Rissen oder Defekten nicht verwenden.
8. Im Sichtfenster überprüfen, ob die Flüssigkeit klar und farblos bis leicht gelblich ist. Das Arzneimittel nicht injizieren, wenn die Flüssigkeit trüb oder verfärbt ist oder Schwebstoffe enthält.
- Die Spritze kann einige kleine Luftblasen enthalten. Das ist normal. Versuchen Sie nicht, diese Luftblasen zu entfernen.

Abbildung F
Nicht verwenden, wenn das Verfalldatum abgelaufen, die Spritze beschädigt oder die Flüssigkeit trüb oder verfärbt ist oder Schwebstoffe enthält. In einem solchen Fall mit Schritt 21 «Enspryng entsorgen» fortfahren und Ihren Arzt bzw. Ihre Ärztin kontaktieren.
Die Spritze aufwärmen lassen
9. Nach dem Überprüfen die Spritze 30 Minuten auf einer sauberen, ebenen Arbeitsfläche (zum Beispiel auf einem Tisch) liegen lassen, damit sie Raumtemperatur annehmen kann (Abbildung G).
Es ist wichtig, dass sich die Spritze leicht erwärmt, da sich das Injizieren des kalten Arzneimittels unangenehm anfühlt und das Drücken des Spritzenkolbens erschwert.
Stets beachten:
- Nichts unternehmen, um den Aufwärmvorgang zu beschleunigen, z. B. durch Verwendung einer Mikrowelle, oder indem die Spritze in warmes Wasser gelegt wird.
- Die Nadelschutzabdeckung nicht abnehmen, während die Spritze Raumtemperatur annimmt.

Abbildung G
Hände waschen
10. Die Hände mit Wasser und Seife waschen (Abbildung H).

Abbildung H
Injektionsstelle wählen
11. Die Injektionsstelle wählen:
- entweder im unteren Teil des Bauchs (Abdomen) oder
- an der Vorderseite und an den Seiten der Oberschenkel (Abbildung I).

Abbildung I
- Zum Injizieren nicht den Bereich von 5 Zentimetern um den Bauchnabel verwenden.
- Nicht in Muttermale, Narben, Blutergüsse oder in Stellen injizieren, an denen die Haut empfindlich, gerötet, verhärtet bzw. nicht intakt ist.
Für jede neue Injektion eine andere Injektionsstelle wählen – die neue Injektionsstelle muss mindestens 2,5 cm von der Stelle entfernt sein, an der Sie zuletzt injiziert haben.
Injektionsstelle reinigen
12. Die Injektionsstelle mit einem Alkoholtupfer abwischen und lufttrocknen lassen.
Stets beachten:
- Die gereinigte Stelle nicht befächern und nicht darauf blasen.
- Die Injektionsstelle nicht erneut berühren, bevor Sie die Injektion verabreichen.

Abbildung J
Enspryng injizieren
13. Den Spritzenzylinder zwischen Daumen und Zeigefinger halten. Mit der anderen Hand die Nadelkappe in einer geraden Linie abziehen. Möglicherweise sehen Sie am Ende der Nadel einen Tropfen Flüssigkeit – dies ist normal und hat keinen Einfluss auf Ihre Dosis (Abbildung K).
- Die Spritze nach dem Entfernen der Kappe innerhalb von 5 Minuten verwenden, da sonst die Nadel verstopfen kann.
Stets beachten:
- Die Nadelkappe erst dann abnehmen, wenn Sie bereit sind, Enspryng zu injizieren.
- Die Nadelkappe nach dem Entfernen nicht wieder aufsetzen, da sonst die Nadel beschädigt werden kann.
- Die Nadel nicht berühren und nicht in Kontakt mit anderen Flächen kommen lassen, nachdem die Nadelkappe entfernt worden ist.

Abbildung K
14. Die Nadelkappe sofort in einen durchstichfesten Entsorgungsbehälter für scharfe und spitze Gegenstände entsorgen. Siehe Schritt 21 «Enspryng entsorgen».
15. Den Spritzenzylinder mit Daumen und Zeigefinger festhalten. Mit der anderen Hand die gereinigte Hautstelle zusammendrücken. (Abbildung L).
16. Die Nadel mit einer schnellen, pfeilartigen Bewegung in einem Winkel zwischen 45° und 90° einführen (Abbildung L).
Stets beachten:
- Die Nadel nicht durch Kleidung hindurch einführen.
- Den Injektionswinkel nicht verändern.
- Die Nadel kein zweites Mal einführen.

Abbildung L
17. Nachdem die Nadel eingeführt worden ist, die zusammengedrückte Haut loslassen.
18. Langsam das gesamte Arzneimittel injizieren, indem der Kolben behutsam vollständig gedrückt wird, bis er die Aktivierungssicherungen berührt (Abbildung M).

Abbildung M
19. Den Kolben vorsichtig loslassen und die Nadel im gleichen Winkel wie beim Einführen aus der Haut gleiten lassen (Abbildung N).

Abbildung N
- Die Nadel ist nun vom Nadelschutz abgedeckt. Wenn die Nadel nicht abgedeckt ist, die Spritze vorsichtig in einen durchstichfesten Entsorgungsbehälter für scharfe und spitze Gegenstände geben, um Verletzungen zu vermeiden. Siehe Schritt 21 «Enspryng entsorgen».
Injektionsstelle versorgen
20. Möglicherweise tritt an der Injektionsstelle etwas Blut aus. Sie können einen Wattebausch oder eine Mullkompresse auf die Injektionsstelle drücken, aber nicht daran reiben. Falls erforderlich, können Sie ein kleines Pflaster auf der Injektionsstelle anbringen. Wenn das Arzneimittel in Kontakt mit der Haut gelangt ist, die betreffende Stelle mit Wasser abwaschen.
Enspryng entsorgen
21. Nicht versuchen, die Kappe wieder auf die Spritze aufzusetzen. Die benutzte Spritze sofort nach Gebrauch in einen Entsorgungsbehälter für scharfe und spitze Gegenstände geben (Abbildung O). Die Spritze nicht in den Haushaltsabfall werfen/im Haushaltsabfall entsorgen und nicht recyceln.

Abbildung O
- Wenden Sie sich an Ihren Arzt bzw. Ihre Ärztin oder an Ihren Apotheker bzw. Ihre Apothekerin, um zu erfahren, wo Sie einen Entsorgungsbehälter für scharfe und spitze Gegenstände erhalten oder welche anderen Arten von durchstichfesten Behältern Sie verwenden können, um Ihre gebrauchten Spritzen und Nadelkappen sicher zu entsorgen, wenn Sie noch keinen solchen Behälter haben.
- Den vollen Entsorgungsbehälter für scharfe und spitze Gegenstände nach den Anweisungen Ihres Arztes bzw. Ihrer Ärztin oder Ihres Apothekers bzw. Ihrer Apothekerin entsorgen.
- Den vollen Entsorgungsbehälter für scharfe und spitze Gegenstände nicht über den Haushaltsabfall entsorgen.
- Den vollen Entsorgungsbehälter für scharfe und spitze Gegenstände nicht recyceln.
▼ Ce médicament fait l'objet d'une surveillance supplémentaire qui permettra l'identification rapide de nouvelles informations relatives à la sécurité. Vous pouvez y contribuer en signalant tout effet secondaire. Voir à la fin de la rubrique «Quels effets secondaires Enspryng peut-il provoquer?» pour savoir comment déclarer les effets secondaires.
Qu’est-ce que l’Enspryng et quand doit-il être utilisé?
Le principe actif d'Enspryng est le satralizumab qui appartient à un groupe de médicaments appelés «anticorps monoclonaux». Les anticorps monoclonaux sont un type de protéine et sont conçus pour reconnaître une substance cible définie et se lier à celle-ci.
Enspryng est destiné au traitement du spectre de la neuromyélite optique (NMOSD) chez l'adulte et l'adolescent.
La NMOSD sont des maladies auto-immunes du système nerveux central (SNC) touchant essentiellement les nerfs optiques et la moelle épinière. L'atteinte des nerfs optiques entraîne des gonflements et des inflammations à l'origine de douleurs et d'une perte de la vision. L'atteinte de la moelle épinière entraîne une faiblesse au niveau des jambes et des bras ou une perte de la capacité à bouger les jambes et les bras, une insensibilité et des troubles de la fonction vésicale et intestinale.
Une rechute ou une «crise» de NMOSD survient en présence d'une inflammation du système nerveux. Chez les personnes atteintes, cette inflammation entraîne de nouveaux symptômes ou des symptômes connus, déjà survenus dans le passé.
Enspryng bloque l'action d'une cytokine appelée interleukine-6 (IL-6), une protéine particulière qui est impliquée dans les processus inflammatoires de l'organisme. Il a été démontré que ce médicament diminue le risque de survenue d'une rechute de NMOSD.
Selon prescription du médecin.
Quand Enspryng ne doit-il pas être pris/utilisé?
Si vous êtes allergique au satralizumab ou à l'un des autres composants du médicament mentionnés à la rubrique «Que contient Enspryng?».
Si vous avez un doute, adressez-vous à votre médecin, à votre pharmacien ou au personnel médical avant d'utiliser Enspryng.
Quelles sont les précautions à observer lors de la prise/de l’utilisation d’Enspryng?
Consultez votre médecin, votre pharmacien ou le personnel médical avant d'utiliser Enspryng si l'un des points suivants vous concerne (ou si vous n'en êtes pas sûr/e):
- Vous présentez une infection. Votre médecin attendra que l'infection ait disparu avant que vous utilisiez Enspryng.
- Vous avez été vacciné/e récemment.
Enspryng peut provoquer des effets secondaires dont vous devez informer votre médecin. Ces effets secondaires sont notamment:
Infections
- Consultez votre médecin avant de recevoir Enspryng si vous pensez que vous présentez une infection. Votre médecin attendra que l'infection ait disparu avant que vous utilisiez Enspryng.
- Informez immédiatement votre médecin ou le personnel médical si vous présentez, pendant ou après le traitement par Enspryng, l'un des signes suivants d'une infection:
- fièvre ou frissons
- toux persistante
- mal de gorge ou nouvelles lésions ouvertes (p. ex. boutons de fièvre).
- Informez votre médecin si vous souffrez ou avez souffert d'une maladie sous-jacente (p.ex. diverticulite, diabète, pneumopathie interstitielle, tuberculose, hépatite B), car celles-ci peuvent favoriser la survenue d'infections.
Vaccinations
- Informez votre médecin si vous avez été vacciné/e récemment ou s'il se peut que vous soyez vacciné prochainement.
- Vous ne devez pas recevoir de vaccins vivants ou de vaccins vivants atténués (p.ex. contre la tuberculose ou la fièvre jaune) pendant le traitement par Enspryng.
- Votre médecin vérifiera si vous avez besoin de vaccins avant de commencer le traitement par Enspryng.
Réactions d'hypersensibilité
- Si vous présentez une réaction d'hypersensibilité pendant ou après l'injection, contactez immédiatement votre médecin (voir «Quels effets secondaires Enspryng peut-il provoquer?»).
- Ne vous administrez pas la dose suivante avant d'avoir informé votre médecin ET que celui-ci vous ait demandé d'administrer la dose suivante.
Enzymes hépatiques
- Enspryng peut augmenter le taux de certains enzymes hépatiques dans votre sang pendant le traitement. Votre médecin fera des analyses de sang pour contrôler ces taux et déterminer la fonction de votre foie.
- Informez immédiatement votre médecin ou le personnel médical si vous présentez, pendant ou après le traitement par Enspryng, l'un des signes suivants d'une augmentation des enzymes hépatiques:
- coloration jaune de la peau ou du blanc des yeux (jaunisse)
- urines foncées
- sensation de malaise et état maladif.
Neutrophiles
Enspryng peut diminuer le nombre de globules blancs particuliers (granulocytes neutrophiles, une forme de cellules immunitaires spécialisées) dans votre sang pendant le traitement. Votre médecin fera des analyses de sang pour contrôler votre nombre de globules blancs.
Tumeurs malignes
- Si vous souffrez d'un cancer, informez-en votre médecin. Votre médecin décidera si un traitement par Enspryng peut néanmoins être envisagé.
Paramètres lipidiques
- Si vous présentez des facteurs de risque cardiovasculaires, tels qu'une hypertension ou des taux trop élevés de cholestérol, informez-en votre médecin. Ces facteurs devront éventuellement être contrôlés pendant le traitement par Enspryng.
Enfants et adolescents
Ce médicament ne doit pas être administré aux enfants de moins de 12 ans, car il n'a pas encore été étudié dans ce groupe d'âge.
Aptitude à la conduite et capacité à utiliser des machines
Aucun effet sur l'aptitude à la conduite et la capacité à utiliser des machines n'a été observé.
Prise d'Enspryng avec d'autres médicaments
Veuillez informer votre médecin ou votre pharmacien si
- vous souffrez d'une autre maladie
- vous êtes allergique ou
- vous prenez déjà d'autres médicaments ou utilisez déjà d'autres médicaments en usage externe (médicaments à usage externe) (même en automédication!).
Enspryng peut-il être pris/utilisé pendant la grossesse ou l’allaitement?
Contraception (femmes)
Les femmes doivent utiliser une méthode contraceptive pendant le traitement et les 5 mois qui suivent la dernière dose d'Enspryng.
Grossesse/Allaitement
Si vous êtes enceinte ou si vous allaitez, si vous pensez être enceinte ou envisagez une grossesse, demandez conseil à votre médecin ou votre pharmacien avant d'utiliser ce médicament.
Vous médecin pourra vous conseiller d'arrêter l'allaitement si vous devez être traitée par Enspryng. On ignore si Enspryng passe dans le lait maternel.
Comment utiliser Enspryng?
Veillez à toujours utiliser ce médicament en suivant exactement les indications de votre médecin ou de votre pharmacien. Demandez conseil à votre médecin ou à votre pharmacien en cas de doute.
Enspryng est administré en injection sous la peau (voie sous-cutanée). Au début, votre médecin ou le personnel infirmier pourront réaliser l'injection d'Enspryng. Votre médecin estimera peut-être que vous pouvez vous injecter vous-même Enspryng. Dans ce cas, vous recevrez une formation appropriée pour apprendre à vous injecter vous-même Enspryng. Adressez-vous à votre médecin ou au personnel médical si vous avez des questions au sujet de l'auto-injection. Des «instructions d'emploi» détaillées figurent à la fin de cette notice d'emballage.
La dose recommandée pour les trois premières injections est de 120 mg (le contenu total de la seringue préremplie) une fois toutes les deux semaines (doses de charge). La première injection sera administrée sous la surveillance de votre médecin ou du personnel médical.
Après administration des doses de charge, la dose recommandée est de 120 mg toutes les quatre semaines (dose d'entretien). Utilisez Enspryng une fois toutes les quatre semaines aussi longtemps que votre médecin vous le prescrit.
La dose recommandée est la même pour tous les patients, aussi pour les adolescents à partir de 12 ans.
Si vous avez utilisé plus d'Enspryng que vous n'auriez dû
Enspryng étant délivré dans une seringue préremplie, il est peu probable que vous en receviez trop. Si toutefois vous êtes inquiet/inquiète, consultez votre médecin, votre pharmacien ou le personnel médical.
Si, par erreur, vous injectez Enspryng plus souvent que ce que vous a dit votre médecin, vous devez contacter votre médecin. Apportez toujours la boîte ou le flacon du médicament, même si ceux-ci sont déjà vides.
Si vous avez oublié d'utiliser Enspryng
Si votre médecin ou le personnel médical vous fait les injections et que vous manquez un rendez-vous, fixez immédiatement un autre rendez-vous.
Si vous vous injectez vous-même Enspryng sans surveillance de votre médecin ou du personnel médical et que vous oubliez une injection, vous devez faire celle-ci dès que possible. N'attendez pas jusqu'à la dose suivante prévue.
Afin que le traitement puisse être pleinement efficace, il est très important que vous continuiez à faire les injections. Après avoir administré la dose oubliée, injectez la dose suivante soit
- après deux semaines (dose de charge) soit
- après quatre semaines (dose d'entretien).
Demandez conseil à votre médecin, à votre pharmacien ou au personnel médical en cas de doute.
Si vous arrêtez d'utiliser Enspryng
N'arrêtez pas au dernier moment d'utiliser Enspryng sans en avoir parlé auparavant avec votre médecin. Si vous avez d'autres questions au sujet de l'utilisation de ce médicament, consultez votre médecin, votre pharmacien ou le personnel médical.
L'utilisation et la sécurité d'Enspryng n'ont pas été établies à ce jour chez les enfants de moins de 12 ans.
Ne changez pas de votre propre chef le dosage prescrit. Adressez-vous à votre médecin ou à votre pharmacien si vous estimez que l'efficacité du médicament est trop faible ou au contraire trop forte.
Quels effets secondaires Enspryng peut-il provoquer?
Comme tous les médicaments, ce médicament peut provoquer des effets secondaires, mais ils ne surviennent pas systématiquement chez tous les patients.
Les effets secondaires suivants ont été rapportés lors de l'utilisation de ce médicament:
Réactions liées à la perfusion
- Les réactions liées à la perfusion sont l'effet secondaire le plus fréquent du traitement par Enspryng (très fréquent: peuvent concerner plus d'une personne sur 10). Il s'agit dans la plupart des cas de réactions légères, mais des réactions graves peuvent parfois aussi survenir.
- Informez immédiatement votre médecin ou le personnel médical si vous présentez des signes ou symptômes d'une réaction liée à la perfusion jusqu'à 24 heures après l'injection. Ces symptômes peuvent notamment être les suivants:
- réaction au site d'injection (rougeur, démangeaisons, douleur)
- maux de tête
- diarrhée
- bouffée de chaleur (bouffée vasomotrice)
- éruption cutanée ou rougeur de la peau
- démangeaisons cutanées
- irritations ou douleurs dans la gorge
- urticaire
- essoufflement
- gonflement de la gorge
- pression artérielle basse (vertiges et étourdissements)
- fièvre
- sensation de fatigue ou de vertiges
- nausées
- battements cardiaques rapides.
Autres effets secondaires:
Informez votre médecin ou le personnel médical si vous remarquez l'un des effets secondaires suivants:
Très fréquent (concerne plus d'un utilisateur sur 10)
- Maux de tête
- Douleurs articulaires
- Diminution du taux de globules blancs dans le sang
- Infections des voies urinaires
- Infections des voies respiratoires
Fréquent (concerne 1 à 10 utilisateurs sur 100)
- Douleurs musculaires
- Raideur
- Migraine
- Sensation d'engourdissement
- Gonflement des jambes, des pieds ou des mains
- Éruption cutanée ou démangeaisons
- Allergies ou rhume des foins
- Diminution du taux de fibrinogène (une protéine impliquée dans la coagulation sanguine) dans le sang
- Augmentation du taux de cholestérol et de triglycérides dans le sang
- Augmentation du taux d'enzymes hépatiques (transaminases) dans le sang
- Coloration jaune de la peau et des yeux (augmentation de la bilirubine dans le sang)
- Diminution du taux de plaquettes dans le sang
- Augmentation du poids corporel
- Grippe
Si vous remarquez des effets secondaires, veuillez en informer votre médecin ou votre pharmacien. Ceci vaut en particulier pour les effets secondaires non mentionnés dans cette notice d'emballage.
À quoi faut-il encore faire attention?
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur la boîte et l'étiquette de la seringue préremplie.
Remarques concernant le stockage
Conserver au réfrigérateur (2-8 °C).
Ne pas agiter. Ne pas congeler.
- Non ouvert, Enspryng peut être conservé hors du réfrigérateur, à température ambiante, en dessous de 30 °C, dans l'emballage extérieur, pendant une durée unique de 8 jours au maximum.
- Après avoir été conservé à température ambiante, le produit doit être utilisé ou éliminé.
Conserver le récipient dans le carton pour le protéger de la lumière.
Conserver hors de portée des enfants.
Enspryng est un liquide incolore à jaune pâle. Vous ne devez pas utiliser ce médicament si le liquide est trouble, coloré ou contient des particules solides en suspension visibles.
Vérifiez que la seringue préremplie et le protège-aiguille ne sont pas endommagés. Ne l'utilisez pas en cas de fissures ou de défaut.
Vous devez commencer l'injection dans les 5 minutes qui suivent le retrait du capuchon protecteur, afin d'éviter que le médicament ne sèche et que l'aiguille ne se bouche. Si la seringue préremplie n'est pas utilisée dans les 5 minutes qui suivent le retrait du capuchon protecteur, vous devez éliminer celle-ci dans un collecteur imperforable pour objets pointus et tranchants, et utiliser une nouvelle seringue préremplie.
Remarques complémentaires
N'éliminez pas de médicaments avec les eaux usées ou avec les déchets ménagers. Demandez à votre pharmacien comment éliminer le médicament lorsque vous ne l'utilisez plus. Vous contribuez ainsi à la protection de l'environnement.
Pour de plus amples renseignements, consultez votre médecin ou votre pharmacien, qui disposent d'une information détaillée destinée aux professionnels.
Que contient Enspryng?
Solution injectable en seringue préremplie.
Principes actifs
Le principe actif est le satralizumab. Chaque seringue préremplie contient 120 mg de satralizumab dans 1 ml.
Excipients
Les autres composants sont: L-histidine, acide L-aspartique, chlorhydrate de L-arginine, poloxamère 188, eau pour préparations injectables.
Numéro d’autorisation
67617 (Swissmedic).
Où obtenez-vous Enspryng? Quels sont les emballages à disposition sur le marché?
En pharmacie, seulement sur ordonnance médicale non renouvelable.
Chaque emballage d'Enspryng contient 1 seringue préremplie.
Titulaire de l’autorisation
Roche Pharma (Suisse) SA, Bâle.
Cette notice d'emballage a été vérifiée pour la dernière fois en juillet 2020 par l'autorité de contrôle des médicaments (Swissmedic).
Ce que vous devez savoir pour utiliser en toute sécurité votre seringue préremplie d’Enspryng
Lisez ces instructions d’emploi:
|
En effet, elles pourraient comporter de nouvelles informations.
|
Remarques importantes
Gardez toujours à l’esprit:
Gardez toujours à l’esprit:
|
Comment conserver la seringue préremplie d’Enspryng?
|
Gardez toujours à l’esprit:
|
Accessoires nécessaires pour l’injection Chaque carton d’Enspryng contient:
Non fournis dans le carton:
|
Seringue préremplie d’Enspryng (Figure A et Figure B) Avant l’utilisation:
Figure A Après utilisation:
Figure B La seringue est munie d’un protège-aiguille qui recouvre automatiquement l’aiguille après la fin de l’injection. |



Préparatifs avant l’utilisation d’Enspryng
1. Sortez du réfrigérateur le carton contenant la seringue et posez-le sur une surface propre et plane (p. ex. une table).
2. Vérifiez la date de péremption au dos du carton (Figure C). Ne l’utilisez pas si la date de péremption figurant sur le carton est dépassée.
3. Vérifiez que le sceau sur l’avant du carton est encore intact (Figure C). Ne l’utilisez pas si le sceau est endommagé.
N’utilisez pas la seringue si la date de péremption est dépassée ou si le sceau n’est plus intact. Dans ce cas, passez à l’étape 21 «Éliminer Enspryng» et contactez votre médecin.

Figure C
4. Ouvrez le carton scellé (Figure D).

Figure D
5. Saisissez avec précaution la seringue au niveau de son corps et sortez-la de son carton (Figure E).
Gardez toujours à l’esprit:
- Ne retournez pas le carton pour sortir la seringue.
- Ne touchez pas le dispositif d’activation du protège-aiguille. Ceci pourrait endommager la seringue.
- Ne tenez pas la seringue au niveau du piston ou du protège-aiguille.

Figure E
Vérifiez la seringue
(Figure F)
6. Vérifiez la date de péremption (EXP) sur la seringue. N’utilisez pas la seringue si cette date est dépassée.
7. Vérifiez que la seringue n’est pas endommagée. Ne l’utilisez pas en cas de fissures ou de défaut.
8. Vérifiez dans la fenêtre que le liquide est limpide et incolore ou légèrement jaunâtre. N’injectez pas le médicament si le liquide est trouble ou coloré, ou contient des particules solides en suspension.
- La seringue peut contenir quelques petites bulles d’air. Cela est normal. N’essayez pas d’éliminer ces bulles d’air.

Figure F
N’utilisez pas la seringue si la date de péremption est dépassée, si la seringue est endommagée ou si le liquide est trouble ou coloré, ou contient des particules solides en suspension. Dans ce cas, passez à l’étape 21 «Éliminer Enspryng» et contactez votre médecin.
Laissez la seringue se réchauffer.
9. Après avoir examiné la seringue, laissez-la reposer 30 minutes sur une surface propre et plane (par exemple une table) pour qu’elle atteigne la température ambiante (Figure G).
Il est important que la seringue se réchauffe légèrement, car lorsque le médicament est froid, l’injection est désagréable et il est plus difficile de pousser le piston de la seringue.
Gardez toujours à l’esprit:
- Ne faites rien pour accélérer le réchauffement, p. ex. en utilisant un four à micro-ondes ou en plongeant la seringue dans de l’eau chaude.
- Ne retirez pas le capuchon protecteur de l’aiguille pendant que la seringue revient à température ambiante.

Figure G
Lavez-vous les mains
10. Lavez-vous les mains avec du savon et de l’eau (Figure H).

Figure H
Choisissez le site de l’injection
11. Choisissez le site d’injection:
- soit dans la partie inférieure du ventre (abdomen) soit
- dans la partie avant ou sur les côtés de la cuisse (Figure I).

Figure I
- Ne faites pas l’injection dans une zone de 5 centimètres autour du nombril.
- Ne faites pas l’injection dans des grains de beauté, des cicatrices, des hématomes ou des endroits où la peau est sensible, rougie, indurée ou n’est pas intacte.
Choisissez un site d’injection différent à chaque nouvelle injection – le nouveau site d’injection doit être distant d’au moins 2,5 cm du dernier site utilisé.
Nettoyez le site de l’injection
12. Essuyez le site d’injection avec un tampon alcoolisé et laissez-le sécher.
Gardez toujours à l’esprit:
- N’éventez pas le site nettoyé ou ne soufflez pas sur celui-ci.
- Ne touchez plus le site d’injection avant de faire l’injection.

Figure J
Injectez Enspryng
13. Tenez le corps de la seringue entre le pouce et l’index. Avec l’autre main, retirez le capuchon de l’aiguille en le tirant dans l’axe. Il se peut que vous voyiez une goutte de liquide à l’extrémité de l’aiguille – cela est normal et n’a aucune incidence sur votre dose (Figure K).
- Utilisez la seringue dans les 5 minutes qui suivent le retrait du capuchon, car l’aiguille risque sinon de se boucher.
Gardez toujours à l’esprit:
- Ne retirez le capuchon de l’aiguille que lorsque vous êtes prêt/e à vous injecter Enspryng.
- Ne remettez pas le capuchon de l’aiguille après l’avoir retiré, vous risqueriez sinon d’endommager l’aiguille.
- Ne touchez pas l’aiguille et ne la mettez pas au contact d’autres surfaces après avoir retiré le capuchon de l’aiguille.

Figure K
14. Éliminez immédiatement le capuchon de l’aiguille dans un collecteur imperforable pour objets pointus et tranchants. Voir l’étape 21 «Éliminer Enspryng».
15. Tenez le corps de la seringue avec le pouce et l’index. Avec l’autre main, pincez la peau nettoyée. (Figure L).
16. Insérez l’aiguille d’un geste rapide, semblable au lancer d’une fléchette, à un angle compris entre 45° et 90° (Figure L).
Gardez toujours à l’esprit:
- N’insérez pas l’aiguille à travers les vêtements.
- Ne modifiez pas l’angle d’injection.
- N’insérez pas l’aiguille une seconde fois.

Figure L
17. Relâchez la peau après avoir inséré l’aiguille.
18. Injectez lentement la totalité du médicament en appuyant doucement et à fond sur le piston jusqu’à ce qu’il touche le dispositif d’activation du protège-aiguille (Figure M).

Figure M
19. Relâchez prudemment le piston et retirez l’aiguille de la peau en maintenant le même angle qu’à l’insertion (Figure N).
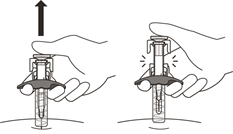
Figure N
- L’aiguille est maintenant recouverte du protège-aiguille. Si l’aiguille n’est pas recouverte, mettez la seringue prudemment dans un collecteur imperforable pour objets pointus et tranchants pour éviter toute blessure. Voir l’étape 21 «Éliminer Enspryng».
Soignez le site d’injection
20. Il se peut qu’un peu de sang s’écoule du site d’injection. Vous pouvez appuyer un tampon d’ouate ou une compresse de gaze sur le site d’injection, mais ne frottez pas. Vous pouvez mettre un petit pansement sur le site d’injection si nécessaire. Si le médicament est entré en contact avec la peau, laver la région concernée à l’eau.
Éliminez Enspryng
21. N’essayez pas de remettre le capuchon sur la seringue. Mettez immédiatement après l’emploi la seringue usagée dans un collecteur pour objets pointus et tranchants (Figure O). Ne jetez/n’éliminez pas la seringue avec les ordures ménagères et ne la recyclez pas.

Figure O
- Adressez-vous à votre médecin ou à votre pharmacien pour savoir où vous procurer un collecteur pour objets pointus et tranchants et quels autres types de récipients imperforables vous pouvez utiliser pour éliminer en toute sécurité vos seringues et capuchons d’aiguille usagés si vous ne disposez pas encore d’un tel récipient.
- Lorsque le collecteur pour objets pointus et tranchants est plein, éliminez-le selon les instructions de votre médecin ou de votre pharmacien.
- N’éliminez pas le collecteur pour objets pointus et tranchants avec les ordures ménagères lorsqu’il est plein.
- Ne recyclez pas le collecteur pour objets pointus et tranchants lorsqu’il est plein.
▼ Questo medicamento è soggetto a monitoraggio addizionale. Ciò consente una rapida identificazione delle nuove conoscenze in materia di sicurezza. Potete contribuire segnalandone gli effetti collaterali. Per istruzioni sulle modalità di notifica degli effetti collaterali si veda al termine del capitolo «Quali effetti collaterali può avere Enspryng?».
Che cos'è Enspryng e quando si usa?
Enspryng contiene il principio attivo satralizumab, che appartiene alla classe di farmaci definiti «anticorpi monoclonali». Gli anticorpi monoclonali sono un tipo di proteine deputate a riconoscere una determinata sostanza bersaglio e a legarvisi.
Enspryng è destinato al trattamento delle malattie dello spettro della neuromielite ottica (neuromyelitis optica spectrum disorders, NMOSD) negli adulti e negli adolescenti.
NMOSD sono malattie autoimmuni del sistema nervoso centrale (SNC), che colpiscono prevalentemente i nervi ottici e il midollo spinale. Le lesioni dei nervi ottici causano gonfiori e infiammazioni che provocano dolore e perdita della capacità visiva. La lesione del midollo spinale determina debolezza nelle gambe e nelle braccia o perdita della capacità di muovere gambe e braccia, o insensibilità e problemi connessi alla funzione della vescica e dell'intestino.
Una recidiva o «attacco» di NMOSD si verifica quando è presente un'infiammazione nel sistema nervoso. Le persone interessate, a causa dell'infiammazione, presentano nuovi sintomi o sintomi noti, già comparsi in precedenza.
Enspryng blocca l'azione di una determinata proteina, una citochina, chiamata interleukina-6 (IL-6) che contribuisce ai processi infiammatori che si svolgono nel corpo. È stato dimostrato che questo medicamento riduce il rischio che compaia una recidiva di NMOSD.
Su prescrizione medica.
Quando non si può usare Enspryng?
Se lei è allergico al satralizumab o ad altri componenti del medicamento, elencati nella rubrica «Cosa contiene Enspryng».
Se non si sente sicuro, prima di usare Enspryng, ne parli con il suo medico, con il suo farmacista o con un operatore sanitario.
Quando è richiesta prudenza nell'uso di Enspryng?
Prima di usare Enspryng, parli con il suo medico, con il suo farmacista o con un operatore sanitario se uno dei seguenti punti la riguarda (o se non è sicuro se uno dei seguenti punti la riguarda):
- Lei è affetto da un'infezione. Il suo medico deve attendere che l'infezione sia guarita prima di poterle prescrivere Enspryng.
- Lei è stato recentemente vaccinato.
Enspryng può causare effetti collaterali che deve comunicare al suo medico. Tra questi vi sono:
Infezioni
- Parli con il suo medico prima di ricevere Enspryng, se pensa di essere affetto da un'infezione. Il suo medico deve attendere che l'infezione sia guarita prima di poter usare Enspryng per lei.
- Informi immediatamente il suo medico o un operatore sanitario, se durante o dopo il trattamento con Enspryng lei presenta uno di questi segni di infezione:
- febbre o brividi
- tosse persistente
- mal di gola o nuove lesioni aperte (p. es. vescicole febbrili).
- Informi il suo medico se soffre o ha sofferto di malattie di base (p. es. diverticolite, diabete, malattie polmonari interstiziali, tubercolosi, epatite B). Queste malattie possono favorire la comparsa di infezioni.
Vaccinazioni
- Informi il suo medico, se lei è stato recentemente vaccinato o se sarà vaccinato in un prossimo futuro.
- Durante il trattamento con Enspryng non deve ricevere vaccini vivi né vaccini vivi attenuati (p. es. contro la tubercolosi o contro la febbre gialla).
- Il suo medico verificherà, prima di iniziare il trattamento con Enspryng, se ha bisogno di vaccinazioni.
Reazioni di ipersensibilità
- Contatti immediatamente il suo medico se durante o dopo l'iniezione si verifica una reazione di ipersensibilità (cfr. la rubrica «Quali effetti collaterali può avere Enspryng?»)
- Si somministri la dose successiva di Enspryng soltanto dopo aver informato il suo medico E questi l'ha invitata a somministrarsi la dose successiva.
Enzimi epatici
- Enspryng può causare un aumento della quantità di enzimi epatici nel suo sangue durante il trattamento. Il suo medico eseguirà esami del sangue per controllare la quantità di questi enzimi e per verificare se il suo fegato funziona bene.
- Informi immediatamente il suo medico o un operatore sanitario, se durante o dopo il trattamento con Enspryng lei presenta uno di questi segni di aumento degli enzimi epatici:
- colorazione gialla della pelle e del bianco dell’occhio (ittero)
- urine scure
- sensazione di malessere e stato di malattia.
Neutrofili
- Enspryng può causare una diminuzione del numero di determinati globuli bianchi (granulociti neutrofili, un tipo di cellule immunitarie specializzate) nel suo sangue durante il trattamento. Il medico eseguirà esami del sangue per controllare il numero dei globuli bianchi.
Tumori maligni
- Se è affetto da un tumore maligno, ne informi il suo medico. Il medico deciderà se si può comunque prendere in considerazione il trattamento con Enspryng.
Parametri lipidici
- Se presenta fattori noti di rischio cardiovascolare (per il cuore e per il circolo), come p. es. pressione alta o valori troppo alti del colesterolo, ne informi il suo medico. Eventualmente, durante il trattamento con Enspryng questi fattori dovranno essere controllati.
Bambini e adolescenti
Questo medicamento non deve essere somministrato a bambini di età inferiore ai 12 anni, perché non è stato ancora studiato in questo gruppo di età.
Effetti sulla guida di veicoli e sulla capacità di utilizzare macchinari
Non sono stati osservati effetti sulla capacità di condurre veicoli o di utilizzare macchine.
Assunzione di Enspryng insieme ad altri medicamenti
Informi il suo medico o il suo farmacista nel caso in cui
- soffre di altre malattie
- soffre di allergie o
- assume altri medicamenti (anche se acquistati di sua iniziativa) o li applica esternamente.
Si può usare Enspryng durante la gravidanza o l'allattamento?
Contraccezione (donne)
Durante il trattamento e per 5 mesi dopo l'assunzione dell'ultima dose di Enspryng, le donne devono impiegare un metodo contraccettivo.
Gravidanza e allattamento
Se è incinta o se allatta, o se ritiene di essere incinta o se sta pianificando una gravidanza, prima di assumere questo medicamento, consulti il suo medico o farmacista.
È possibile che il suo medico le consigli di interrompere l'allattamento, se dovrà essere trattata con Enspryng. Non è noto se Enspryng passi nel latte materno.
Come usare Enspryng?
Usi questo medicamento sempre esattamente come prescritto dal suo medico o dal suo farmacista. Se ha dubbi, chieda al suo medico o al suo farmacista.
Enspryng viene somministrato sotto forma di siringa (iniezione) sotto la pelle (sottocutanea). Inizialmente, l'iniezione di Enspryng può essere somministrata dal medico o dall'operatore sanitario. Il suo medico potrebbe ritenere che lei sia in grado di iniettarsi Enspryng da solo. In questo caso, sarà istruito su come somministrarsi da solo l'iniezione di Enspryng. Se ha delle domande sull'autoiniezione, ne parli con il suo medico o con l'operatore sanitario. Troverà esaurienti «Istruzioni per l'uso» alla fine di questo foglietto illustrativo.
Per le prime tre iniezioni la dose raccomandata è di 120 mg (tutto il contenuto della siringa preriempita) una volta ogni due settimane (dosi di saturazione). La prima iniezione sarà iniettata sotto la supervisione del medico o dell'operatore sanitario.
Dopo la somministrazione delle dosi di saturazione, la dose raccomandata è di 120 mg ogni quattro settimane (dose di mantenimento). Continui a somministrarsi Enspryng una volta ogni 4 settimane per tutto il tempo che il suo medico riterrà necessario.
La dose raccomandata è uguale per tutti i pazienti, anche per gli adolescenti a partire dai 12 anni.
Se ha utilizzato una quantità di Enspryng maggiore del dovuto
Poiché Enspryng viene somministrato con una siringa preriempita, è improbabile che riceva una dose troppo alta. Se comunque fosse preoccupato, si rivolga al suo medico, al suo farmacista o a un operatore sanitario.
Se per errore si inietta Enspryng più volte di quelle prescritte dal suo medico, lo consulti, portando con sé la scatola o il flaconcino con il medicamento, anche se vuoti.
Se ha dimenticato di usare Enspryng
Se lei riceve le sue iniezioni dal medico o da un operatore sanitario e perde un appuntamento, concordi immediatamente un nuovo appuntamento.
Se lei si inietta Enspryng da solo e non sotto la supervisione del medico o di un operatore sanitario e dimentica un'iniezione, dovrebbe recuperarla quanto prima, senza aspettare la successiva dose programmata.
Affinché il trattamento possa essere pienamente efficace, è molto importante proseguire con le iniezioni. Dopo essersi iniettata la dose dimenticata, si inietti la dose successiva o
- dopo due settimane (dose di saturazione) o
- dopo quattro settimane (dosi di mantenimento).
Se ha dubbi chieda al medico o al farmacista o all'operatore sanitario.
Se interrompe l'uso di Enspryng
Non interrompa improvvisamente l'uso di Enspryng senza averne prima parlato con il suo medico. Se ha ulteriori domande sull'impiego del medicamento si rivolga al suo medico, al suo farmacista o a un operatore sanitario.
L'uso e la sicurezza di Enspryng nei bambini sotto i 12 anni finora non sono stati esaminati.
Non modifichi di propria iniziativa la posologia prescritta. Se ritiene che l'azione del medicamento sia troppo debole o troppo forte, ne parli al suo medico o al suo farmacista.
Quali effetti collaterali può avere Enspryng?
Come tutti i medicamenti, anche questo medicamento può avere effetti collaterali, che tuttavia non compaiono necessariamente in ogni paziente.
Durante l'uso di questo medicamento sono stati riportati i seguenti effetti collaterali:
Reazioni correlate all'iniezione
- Le reazioni correlate all'iniezione rappresentano gli effetti collaterali più frequenti del trattamento con Enspryng (molto comuni, possono riguardare più di 1 persona su 10). Nella maggior parte dei casi si tratta di reazioni lievi, talora tuttavia possono anche comparire reazioni gravi.
- Informi immediatamente il suo medico o un operatore sanitario se entro 24 ore dopo l'iniezione dovessero comparire segni o sintomi di una reazione correlata all'iniezione. Possono comparire, tra gli altri, i seguenti sintomi:
- una reazione nel punto dell'iniezione (arrossamento, prurito, dolori)
- mal di testa
- diarrea
- vampate di calore (flushing)
- eruzione o arrossamento cutaneo
- pelle pruriginosa
- irritazione o dolori alla faringe
- orticaria
- affanno
- gonfiore alla faringe
- pressione del sangue bassa (vertigine e stordimento)
- febbre
- sensazione di stanchezza o di vertigine
- nausea
- battito cardiaco rapido.
Altri effetti collaterali:
informi immediatamente il suo medico o un operatore sanitario se osserva uno dei seguenti effetti collaterali:
Molto comune (riguarda più di 1 su 10 utilizzatori)
- Mal di testa
- Dolori articolari
- Concentrazione ridotta dei globuli bianchi
- Infezioni delle vie urinarie
- Infezioni delle vie respiratorie
Comune (riguarda da 1 a 10 utilizzatori su 100)
- Dolori muscolari
- Rigidità
- Emicrania
- Intorpidimento
- Gonfiore alle gambe, ai piedi o alle mani
- Eruzione cutanea e prurito
- Allergie o rinite da fieno
- Bassi livelli di fibrinogeno nel sangue (una proteina che partecipa alla coagulazione del sangue)
- Aumento della concentrazione del colesterolo e dei trigliceridi nel sangue
- Aumento della concentrazione degli enzimi epatici (transaminasi) nel sangue
- Pelle e occhi giallastri (bilirubina aumentata nel sangue)
- Riduzione della concentrazione piastrinica nel sangue
- Aumento del peso corporeo
- Influenza
Se osserva effetti collaterali, si rivolga al suo medico o al suo farmacista, soprattutto se si tratta di effetti collaterali non descritti in questo foglietto illustrativo.
Di che altro occorre tener conto?
Il medicamento non deve essere utilizzato oltre la data indicata con «EXP» sulla scatola pieghevole e sull'etichetta della siringa preriempita.
Indicazione di stoccaggio
Conservare in frigorifero (2-8 °C).
Non agitare. Non congelare.
- Enspryng può essere conservato in confezione integra nel suo cartone, per una sola volta, fino a 8 giorni, a temperatura ambiente non superiore ai 30 °C fuori dal frigorifero.
- Dopo la conservazione a temperatura ambiente il preparato dovrà essere utilizzato o smaltito.
Conservare il contenitore nel suo cartone per tenere il contenuto al riparo dalla luce.
Tenere fuori dalla portata dei bambini.
Enspryng è un liquido incolore o giallo pallido. Non deve utilizzare questo medicamento se constata un aspetto torbido, una colorazione anomala o se sono visibili sostanze in sospensione.
Controlli la siringa preriempita e la protezione dell'ago per riscontrare eventuali danni. Non usare se sono presenti fessure o difetti.
Dopo aver rimosso il cappuccio di protezione, l'iniezione deve essere iniziata entro 5 minuti per impedire che il medicamento si secchi e che l'ago si ostruisca. Se la siringa preriempita non viene usata entro 5 minuti dalla rimozione del cappuccio di protezione, essa deve essere smaltita in un contenitore di smaltimento per oggetti taglienti o appuntiti a prova di perforazione e bisogna usare una nuova siringa preriempita.
Ulteriori indicazioni
Non smaltire i medicamenti nelle acque di scarico né nei rifiuti domestici. Chieda al suo farmacista come smaltire il medicamento quando non lo utilizza più. In questo modo, contribuirà a proteggere l'ambiente.
Il medico o il farmacista, che sono in possesso di un'informazione professionale dettagliata, possono darle ulteriori informazioni.
Cosa contiene Enspryng?
Soluzione iniettabile in una siringa preriempita.
Principi attivi
Il principio attivo è satralizumab. Ciascuna siringa preriempita contiene 120 mg di satralizumab in 1 ml.
Sostanze ausiliarie
Gli ulteriori componenti sono L-istidina, acido L-asparaginico, L-arginina cloridrato, polossamero 188, acqua per preparazioni iniettabili.
Numero dell’omologazione
67617 (Swissmedic).
Dove è ottenibile Enspryng? Quali confezioni sono disponibili?
In farmacia solo dietro presentazione della prescrizione medica non rinnovabile.
Ogni confezione di Enspryng contiene 1 siringa preriempita.
Titolare dell’omologazione
Roche Pharma (Svizzera) SA, Basilea.
Questo foglietto illustrativo è stato controllato l'ultima volta nel luglio 2020 dall'autorità competente in materia di medicamenti (Swissmedic).
Ciò che deve sapere per usare la sua siringa preriempita Enspryng in modo sicuro
Legga queste istruzioni per l’uso:
|
Il motivo di questo è che potrebbero contenere informazioni aggiornate.
|
Avvertenze importanti
Tenere sempre presente:
Tenere sempre presente:
|
Come conservare la siringa preriempita Enspryng?
|
Tenere sempre presente:
|
Accessori necessari per l’iniezione Ogni confezione di Enspryng contiene:
Nella confezione non sono contenuti:
|
Enspryng siringa preriempita (Figura A e Figura B) Prima dell’uso:
Figura A Dopo l’uso:
Figura B La siringa dispone di una protezione per l’ago che la copre automaticamente dopo l’iniezione. |



Preparazione per l’uso di Enspryng
1. Prelevare il cartone con la siringa dal frigorifero e posarla su un piano di lavoro pulito, piano (p. es. un tavolo).
2. Controllare la data di scadenza sul retro della confezione (Figura C). Non usare se è stata superata la data di scadenza indicata sulla confezione.
3. Controllare se il sigillo sul lato anteriore del cartone è ancora integro (Figura C). Non eseguire l'iniezione se il sigillo è danneggiato.
Non usare se è stata superata la data di scadenza o se il sigillo non è più integro. In uno di questi casi proseguire con fase 21 «smaltire Enspryng» e contattare il suo medico.

Figura C
4. Aprire il cartone sigillato (Figura D).

Figura D
5. Afferrare con prudenza la siringa dalla parte del cilindro e prelevarla dal cartone (Figura E).
Tenere sempre presente:
- Non capovolgere il cartone per prelevare la siringa.
- Non toccare i dispositivi di sicurezza dell’attivazione. La siringa potrebbe venirne danneggiata.
- Non afferrare la siringa dalla parte dello stantuffo e dalla parte della protezione dell’ago.

Figura E
Controllare la siringa
(Figura F)
6. Controllare la data di scadenza (EXP) sulla siringa. Non usare la siringa se è scaduta.
7. Controllare se la siringa è danneggiata. Non usare se sono presenti fessure o difetti.
8. Controllare nella finestrella d’ispezione se il liquido è limpido e incolore o giallognolo. Non effettui l'iniezione se la soluzione è torbida, ha un colore anomalo o contiene particelle in sospensione.
- La siringa può contenere alcune piccole bolle d’aria, è normale. Non cerchi di rimuovere queste bolle d’aria.
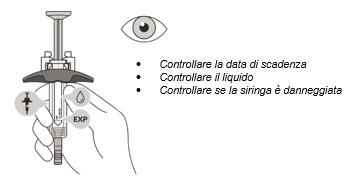
Figura F
Non usare se la data di scadenza è superata, se la siringa è danneggiata o se il liquido è torbido, ha un colore anomalo o contiene particelle in sospensione. In uno di questi casi proseguire con fase 21 «Smaltire Enspryng» e contattare il medico.
Fare riscaldare la siringa
9. Dopo aver effettuato tutti i controlli, lasciare la siringa per 30 minuti su un piano di lavoro pulito, piano (per esempio su un tavolo) perché possa raggiungere la temperatura ambiente (Figura G).
È importante che la siringa si riscaldi lievemente, infatti l’iniezione del medicamento freddo dà una sensazione sgradevole e rende più difficile premere lo stantuffo.
Tenere sempre presente:
- Non fare nulla per accelerare il processo di riscaldamento, p. es. scaldando la siringa nel forno a microonde, o appoggiandola nell’acqua calda.
- Non rimuovere la copertura di protezione dell’ago mentre la siringa raggiunge la temperatura ambiente.

Figura G
Lavarsi le mani
10. Lavarsi le mani con acqua e sapone (Figura H).

Figura H
Scegliere il punto di iniezione
11. Scegliere il punto di iniezione:
- o nella parte inferiore della pancia (addome) o
- sulla faccia anteriore o sui lati delle cosce (Figura I).

Figura I
- Non usare per l’iniezione la zona di 5 centimetri intorno all’ombelico.
- Non iniettare in nei, in cicatrici, in ecchimosi o in sedi nelle quali la pelle è sensibile, arrossata, indurita o comunque non integra.
Per ogni nuova iniezione scegliere un nuovo punto di iniezione – il nuovo punto di iniezione deve essere distante almeno 2,5 cm dal punto in cui ha fatto l’iniezione precedente.
Pulire il punto di iniezione
12. Pulire il punto d’iniezione con un tampone imbevuto di alcoole lasciarlo asciugare all’aria.
Tenere sempre presente:
- Non ventilare il punto pulito né soffiarci su.
- Non toccare più il punto di iniezione prima di somministrare l’iniezione.

Figura J
Iniettare Enspryng
13. Tenere il cilindro della siringa tra pollice e indice. Con l’altra mano togliere il cappuccio dell’ago tenendolo dritto. È possibile che all’estremità dell’ago veda una goccia di liquido - ciò è normale e non ha nessuna influenza sulla sua dose (Figura K).
- Dopo la rimozione del cappuccio, usare la siringa entro 5 minuti, altrimenti l’ago potrebbe ostruirsi.
Tenere sempre presente:
- Rimuovere il cappuccio dell’ago soltanto quando è pronto a iniettarsi Enspryng.
- Dopa aver rimosso il cappuccio, non riposizionarlo in sede altrimenti l’ago potrebbe venirne danneggiato.
- Non toccare l’ago e non farlo venire a contatto con altre superfici dopo aver rimosso il cappuccio.

Figura K
14. Smaltire subito il cappuccio dell’ago in un contenitore per oggetti taglienti e appuntiti a prova di perforazione. Vedi fase 21 «Smaltire Enspryng».
15. Tenere il cilindro della siringa tra pollice e indice. Con l’altra mano comprimere il punto della pelle precedentemente pulito (Figura L).
16. Introdurre l’ago con un movimento rapido, a freccia, con un angolo tra 45° e 90° (Figura L).
Tenere sempre presente:
- Non introdurre l’ago attraverso i vestiti.
- Non cambiare l’angolo di iniezione.
- Non introdurre l’ago una seconda volta.

Figura L
17. Dopo aver introdotto l’ago, rilasciare la pelle compressa.
18. Iniettare lentamente tutto il medicamento, premendo completamente lo stantuffo con delicatezza fino a quando non tocchi i dispositivi di sicurezza per l’attivazione (Figura M).

Figura M
19. Rilasciare con prudenza lo stantuffo ed estrarre l’ago dalla pelle, mantenendo lo stesso angolo dell’introduzione (Figura N).

Figura N
- L’ago è ora coperto dalla protezione dell’ago. Se l’ago non è coperto, per evitare di ferirsi, gettare la siringa con prudenza in un contenitore per lo smaltimento per oggetti taglienti e appuntiti a prova di perforazione. Vedi fase 21 «Smaltire Enspryng».
Medicare il punto di iniezione
20. È possibile che dal punto di iniezione fuoriesca un po’ di sangue. Può premere un batuffolo di cotone o una compressa di garza sul punto di iniezione, senza strofinarlo. Se necessario, può applicare un piccolo cerotto sul punto di iniezione. Se il medicamento è venuto a contatto con la pelle, lavare con acqua la sede interessata.
Smaltire Enspryng
21. Non tentare di rimettere il cappuccio sulla siringa. Gettare la siringa usata subito dopo l’uso in un contenitore per lo smaltimento di oggetti taglienti e appuntiti a prova di perforazione (Figura O). Non gettare né smaltire la siringa nei rifiuti domestici e non riciclarla.

Figura O
- Si rivolga al suo medico o al suo farmacista per sapere dove può trovare un contenitore per lo smaltimento di oggetti taglienti e appuntiti o quali altri tipi di contenitore a prova di perforazione può usare per smaltire le siringhe usate e i cappucci in modo sicuro, qualora non ne avesse uno simile a disposizione.
- Smaltire il contenitore per oggetti taglienti e appuntiti una volta pieno secondo le istruzioni del suo medico o del suo farmacista.
- Non smaltire il contenitore per oggetti taglienti e appuntiti una volta pieno insieme ai rifiuti domestici.
- Non riciclare il contenitore, una volta pieno, per oggetti taglienti e appuntiti.
▼ Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, den Verdacht einer neuen oder schwerwiegenden Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Rubrik «Unerwünschte Wirkungen».
Zusammensetzung
Wirkstoffe
Satralizumabum (gentechnologisch hergestellt unter Verwendung von CHO [Chinese Hamster Ovary]-Zellen).
Hilfsstoffe
L-histidinum; acidum L-asparticum; L-arginini hydrochloridum; poloxamera 188; aqua ad iniectabilia q.s. ad solutionem pro 1,0 ml.
Darreichungsform und Wirkstoffmenge pro Einheit
Injektionslösung in einer Fertigspritze.
Gebrauchsfertige sterile Lösung zur subkutanen (s. c.) Injektion in einer Fertigspritze mit Nadelschutzvorrichtung (Needle Safety Device, NSD), die eine Einzeldosis enthält.
Enspryng Lösung zur s. c. Injektion ist eine farblose bis hellgelbe Flüssigkeit in einer Fertigspritze mit 1 ml Lösung. Jede Fertigspritze enthält 120 mg Satralizumab.
Indikationen/Anwendungsmöglichkeiten
Enspryng wird als Monotherapie oder in Kombination mit einer Immunsuppressionstherapie (IST) zur Behandlung von Neuromyelitis-Optica-Spektrum-Erkrankungen (NMOSD) bei Erwachsenen und Jugendlichen angewendet, bei denen Aquaporin-4-IgG-Antikörper nachweisbar sind (d.h. die AQP4-IgG-seropositiv sind).
Dosierung/Anwendung
Allgemein
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Empfohlene Dosierung
Enspryng muss als subkutane Injektion verabreicht werden.
Therapieeinleitung/Aufsättigungsdosis
Die empfohlene Aufsättigungsdosis beträgt 120 mg per s. c. Injektion alle 2 Wochen (erste Dosis in Woche 0, zweite Dosis in Woche 2 und dritte Dosis in Woche 4) bei den ersten drei Anwendungen.
Erhaltungsdosis
Die empfohlene Erhaltungsdosis beträgt 120 mg per s. c. Injektion alle 4 Wochen (ab Woche 8).
Therapiedauer
Enspryng ist für die Langzeitbehandlung vorgesehen.
Dosisanpassung aufgrund unerwünschter Wirkungen/Interaktionen
Veränderungen der Leberenzyme
Wenn die Erhöhung der Alanin-Aminotransferase (ALT) oder Aspartat-Transaminase (AST) über dem 5-Fachen der oberen Normgrenze (ONG) liegt und mit einer Erhöhung des Bilirubinspiegels einhergeht, muss die Behandlung mit Enspryng dauerhaft beendet werden.
Liegt die Erhöhung des ALT- oder AST-Werts über dem 5-Fachen der oberen Normgrenze (ONG), jedoch nicht verbunden mit einer Erhöhung des Bilirubinspiegels, sollte die Behandlung mit Enspryng beendet werden. Sie kann fortgesetzt werden (120 mg per s. c. Injektion alle 4 Wochen), wenn die ALT- und AST-Werte wieder im Normalbereich liegen, sowie auf der Grundlage einer Einschätzung des Verhältnisses zwischen Nutzen und Risiko der Behandlung des Patienten. Wenn entschieden wird, die Behandlung fortzusetzen, müssen die Leberwerte engmaschig überwacht werden. Falls danach ein erneuter Anstieg der ALT/AST und/oder von Bilirubin festgestellt wird, muss das Arzneimittel dauerhaft abgesetzt werden.
Neutropenie
Wenn die Neutrophilenzahl weniger als 1,0 x 109/l beträgt und in mehreren Tests bestätigt wird, sollte Enspryng abgesetzt werden, bis die Neutrophilenzahl wieder über 1,0 x 109/l gestiegen ist.
Kombinationstherapie
Enspryng kann als Monotherapie oder in Kombination mit oralen Kortikosteroiden (OKs), Azathioprin (AZA) oder Mycophenolatmofetil (MMF) angewendet werden (siehe Rubrik «Eigenschaften/Wirkungen, klinische Wirksamkeit»). Es sind ferner die Fachinformationen zu diesen Präparaten zu beachten.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Die Sicherheit und Wirksamkeit von Enspryng bei Patienten mit Leberfunktionsstörung wurden nicht untersucht (siehe Rubrik «Pharmakokinetik, Kinetik spezieller Patientengruppen»).
Patienten mit Nierenfunktionsstörungen
Die Sicherheit und Wirksamkeit von Enspryng bei Patienten mit Nierenfunktionsstörung wurden nicht formal untersucht. Da Enspryng jedoch ein monoklonaler Antikörper ist, der im Körper Abbauprozessen unterliegt (und nicht durch Ausscheidung über die Nieren entfernt wird), ist bei Patienten mit Nierenfunktionsstörung voraussichtlich keine Dosisanpassungen erforderlich (siehe Rubrik «Pharmakokinetik, Kinetik spezieller Patientengruppen»). Patienten mit leichter Nierenfunktionsstörung wurden in klinische Studien eingeschlossen, und bei diesen Patienten war die Pharmakokinetik von Satralizumab nicht beeinflusst (siehe Rubrik «Pharmakokinetik, Kinetik spezieller Patientengruppen»).
Ältere Patienten
Die Sicherheit und Wirksamkeit von Enspryng wurden bei älteren Patienten im Alter bis zu 74 Jahren untersucht. Bei Patienten ≥65 Jahren sind keine Dosisanpassungen erforderlich (siehe Rubrik «Pharmakokinetik, Kinetik spezieller Patientengruppen»).
Die Sicherheit und Wirksamkeit von Enspryng bei älteren Patienten im Alter >74 Jahren wurden nicht untersucht (siehe Rubrik «Dosierung/Anwendung, Spezielle Dosierungsanweisungen»).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Enspryng wurden bei einer begrenzten Anzahl (N=4) von jugendlichen Patienten im Alter von ≥12 Jahren untersucht. Die Ergebnisse zur Pharmakokinetik, Wirksamkeit und Sicherheit waren mit den bei Erwachsenen erzielten Ergebnissen vergleichbar (siehe Rubrik «Eigenschaften/Wirkungen, klinische Wirksamkeit» und Rubrik «Pharmakokinetik, Kinetik spezieller Patientengruppen»).
Die Sicherheit und Wirksamkeit von Enspryng bei pädiatrischen Patienten im Alter <12 Jahren wurden nicht untersucht.
Verspätete oder ausgelassene Dosisgabe
Falls eine Injektion ausgelassen wird, sollte diese so schnell wie möglich nachgeholt werden und nicht die nächste planmässige Dosis abgewartet werden. Nach der Gabe der verspäteten oder ausgelassenen Dosis sollte zwischen den Dosen das Behandlungsintervall von 2 Wochen (Aufsättigungsphase) bzw. 4 Wochen (Erhaltungsphase) beibehalten werden.
Verabreichungsschema
Die erste Injektion muss unter Aufsicht einer qualifizierten medizinischen Fachperson erfolgen. Ein erwachsener Patient/eine Betreuungsperson kann nachfolgende Injektionen mit Enspryng zu Hause verabreichen, wenn der behandelnde Arzt dies für angemessen hält und der erwachsene Patient/die Betreuungsperson die Injektionstechnik beherrscht.
Patienten/Betreuungspersonen sollten sofort einen Arzt hinzuziehen, wenn der Patient Symptome einer schwerwiegenden allergischen Reaktion entwickelt, und sich bei dem behandelnden Arzt erkundigen, ob die Behandlung mit Enspryng fortgesetzt werden kann oder nicht.
Art der Anwendung
Subkutane (s. c.) Injektion
Enspryng muss als subkutane Injektion verabreicht werden.
Die empfohlenen Injektionsstellen sind die Bauchdecke und die Oberschenkel. Die Injektionsstellen sollten gewechselt werden, und die Injektionen sollten auf keinen Fall in Muttermale, Narben oder in Bereiche verabreicht werden, an denen die Haut druckempfindlich, verletzt, gerötet, verhärtet oder nicht intakt ist.
Eine umfassende Anleitung zur Verabreichung von Enspryng sind in den «Hinweisen für die Handhabung» enthalten.
Kontraindikationen
Enspryng ist bei Patienten mit bekannter Überempfindlichkeit auf Satralizumab oder einen der Hilfsstoffe kontraindiziert.
Warnhinweise und Vorsichtsmassnahmen
Infektionen
Bei Patienten mit aktiver Infektion ist die Gabe von Enspryng auf einen späteren Zeitpunkt zu verschieben, bis die Infektion abgeklungen ist (siehe Rubrik «Dosierung/Anwendung, Verspätete oder ausgelassene Dosen»). Patienten mit wiederkehrenden Infektionen oder Patienten, die an Grunderkrankungen leiden, die das Auftreten von Infektionen begünstigen (z.B. Divertikulitis, Diabetes und interstitielle Lungenerkrankung) sollten mit Vorsicht behandelt werden.
Immunsuppression
Unter Enspryng kann die humorale Immunantwort beeinträchtigt werden.
Tuberkulose
Wie auch bei anderen immunmodulatorischen Therapien empfohlen, sollten alle Patienten vor Beginn einer Therapie mit Enspryng auf das Vorliegen einer latenten Tuberkuloseinfektion untersucht werden. Patienten mit einer latenten Tuberkulose sollten vor Beginn einer Therapie mit Enspryng mit antimykobakterieller Standardtherapie behandelt werden.
Impfungen
Lebendimpfstoffe oder attenuierte Lebendimpfstoffe sollten nicht gleichzeitig mit Enspryng verabreicht werden, da die klinische Sicherheit nicht erwiesen ist. Das Zeitintervall zwischen der Verabreichung von Lebendimpfstoffen und der Einleitung der Enspryng-Therapie sollte den gegenwärtigen Impfrichtlinien bezüglich immunmodulatorischer/immunsuppressiver Wirkstoffe entsprechen.
Es liegen keine Daten über die Auswirkungen einer Impfung bei Patienten vor, die Enspryng erhalten. Es wird empfohlen, bei allen Patienten sämtliche Impfungen in Übereinstimmung mit den gegenwärtigen Impfrichtlinien auf den aktuellen Stand zu bringen, bevor eine Therapie mit Enspryng eingeleitet wird.
Leberenzyme
Während der Behandlung mit Enspryng wurden leichte und mässige Erhöhungen der Lebertransaminasen beobachtet, die überwiegend unter dem 5-Fachen der ONG lagen, nicht behandlungsbeschränkend und während der Gabe von Enspryng reversibel waren.
Die ALT- und AST-Werte sollten in den ersten 3 Behandlungsmonaten alle 4 Wochen kontrolliert werden, anschliessend 1 Jahr lang alle 3 Monate und danach der klinischen Indikation entsprechend. Bzgl. Empfehlungen zur Beendigung der Behandlung siehe Rubrik «Dosierung/Anwendung, Dosisanpassungen aufgrund unerwünschter Wirkungen».
Hepatitis B Reaktivierung
Im Zusammenhang mit biologischen Therapien wurde über eine Reaktivierung von Hepatitis B berichtet. In klinischen Studien mit Enspryng wurden Patienten, die positiv auf Hepatitis getestet wurden, von der Teilnahme ausgeschlossen.
Neutrophilenzahl
Infolge der Behandlung mit Enspryng ist es zu verringerten Neutrophilenzahlen gekommen. Bei 9.6% unter Enspryng und bei 5.4% der Patienten unter Placebo trat eine Neutropenie vom Grad 3 und 4 auf (siehe Rubrik «Unerwünschte Wirkungen»).
Die Neutrophilenzahl sollte 4 bis 8 Wochen nach Beginn der Behandlung und je nach klinischer Indikation auch danach überwacht werden. Empfehlungen im Hinblick auf die Unterbrechung der Therapie: siehe Rubrik «Dosierung/Anwendung».
Arzneimittelmissbrauch und –abhängigkeit
Es wurden keine Studien zu Arzneimittelmissbrauch oder -abhängigkeit durchgeführt. Die verfügbaren Daten lassen jedoch nicht erkennen, dass die Behandlung mit Enspryng zu einer Abhängigkeit führt.
Bei Umstellung der Therapie auf eine andere Darreichungsform und/oder ein anderes Arzneimittel mit gleichem Wirkstoff ist Vorsicht geboten. Der Patient sollte adäquat kontrolliert werden.
Malignome
Immunmodulierende Arzneimittel können das Risiko für Malignitäten erhöhen.
Die Auswirkung einer Behandlung mit Enspryng auf die Entwicklung von malignen Erkrankungen ist nicht bekannt.
Überempfindlichkeitsreaktionen
Bei Patienten unter Behandlung mit Biologika können Überempfindlichkeitsreaktionen auftreten. Vor Therapiebeginn sind die Patienten über mögliche Symptome einer Überempfindlichkeitsreaktion zu informieren.
Die erste Injektion muss unter Aufsicht einer qualifizierten medizinischen Fachperson erfolgen.
Bei Auftreten von Überempfindlichkeitsreaktionen müssen die Patienten ihren Arzt sofort informieren und gegebenenfalls notfallmässig aufsuchen.
Kardiovaskuläre Ereignisse
IL-6 Rezeptor Inhibitoren können das Risiko für kardiovaskuläre Erkrankungen erhöhen. Insbesondere Patienten mit Risikofaktoren wie arterielle Hypertonie, Dyslipidämie und Diabetes mellitus sollten daher regelmässig kontrolliert werden (EKG, Blutdruckmessung).
Lipidparameter
Erhöhungen der Triglyceride und des Cholesterins (Lipidparameter) wurden bei mit Enspryng behandelten Patienten beobachtet (siehe «Unerwünschte Wirkungen»). Patienten mit erhöhten Lipidparametern sollten entsprechend den aktuellen klinischen Richtlinien für die Behandlung von Hyperlipidämie behandelt werden.
Aktivierung des Komplementsystems
Bei einer Behandlung mit IL-6 Rezeptor Inhibitoren kann es zu einer Aktivierung des Komplementsystems kommen. Basierend auf den bisherigen klinischen Daten konnte ein derartiger Effekt unter Enspryng nicht beobachtet werden. Auf Grund der limitierten Daten kann das diesbezüglich Risiko für Enspryng jedoch nicht sicher beurteilt werden.
Demyelinisierende ZNS Erkrankungen
Bei einer Behandlung mit IL-6 Rezeptor Inhibitoren wurde das Auftreten von anderen entzündlichen ZNS Erkrankungen beobachtet. Es sollte auf Symptome geachtet werden, die auf eine neu auftretende demyelinisierende Erkrankung des ZNS hinweisen. Auf Grund der limitierten Daten kann das diesbezügliche Risiko für Enspryng jedoch nicht sicher beurteilt werden.
Divertikel-/Darm-Perforation
Bei einer Behandlung mit IL-6 Rezeptor Inhibitoren von Patienten mit rheumatoider Arthritis (RA) wurde das Auftreten von Divertikel- und Darm-Perforationen beobachtet. In den pivotalen Studien mit Enspryng wurden Patienten mit anamnestisch bekannter Divertikulitis ausgeschlossen. Ein erhöhtes Risiko für Divertikel-/Darm-Perforation wie dies bei anderen Inhibitoren des IL-6 Rezeptors auftritt, ist unter der Behandlung mit Enspryng nicht auszuschliessen. Enspryng sollte bei Patienten mit intestinaler Ulzeration oder Divertikulitis in der Anamnese nur mit Vorsicht angewendet werden. Bei Auftreten akuter Bauchschmerzen sollten die Patienten unverzüglich untersucht werden, damit eine gastrointestinale Perforation frühzeitig erkannt werden kann.
Interaktionen
Mit Enspryng wurden keine formalen Arzneimittelinteraktionsstudien durchgeführt.
Pharmakokinetische Interaktionen
Die Expression hepatischer CYP450-Enzyme wird durch Zytokine wie z.B. IL-6, die chronische Entzündungen stimulieren, unterdrückt. Daher könnte sich die Expression von CYP450 verändern, wenn eine Zytokinhemmung mit Enspryng eingeleitet wird.
Daher sollten Patienten, die Arzneimittel einnehmen, deren Dosis individuell eingestellt wird und die durch CYP450 3A4, 1A2 oder 2C9 metabolisiert werden (z.B. Atorvastatin, Kalziumkanalblocker, Theophyllin, Warfarin, Phenytoin, Cyclosporin oder Benzodiazepine), zu Beginn und Ende einer Therapie mit Enspryng kontrolliert werden und die Dosis dieser Substanzen bei Bedarf angepasst werden. In Anbetracht seiner langen Eliminationshalbwertszeit kann die Wirkung von Enspryng auf die Aktivität von CYP450-Enzymen nach Beendigung der Behandlung über mehrere Wochen bestehen bleiben.
In Analysen der Populationspharmakokinetik wurden keine Auswirkungen von AZA, Kortikosteroiden oder MMF auf die Clearance von Enspryng festgestellt.
Schwangerschaft/Stillzeit
Schwangerschaft
Es liegen keine Daten zur Anwendung von Enspryng bei Schwangeren vor.
Studien bei Affen zeigten keine direkte Toxizität mit Auswirkung auf Schwangerschaft, Entwicklung des Föten und postnatale Entwicklung. Bei Tieren passiert Satralizumab die Plazentaschranke und bei Nachkommen von Tieren, die mit Satralizumab behandelt worden waren, standen einige Befunde möglicherweise mit der pharmakologischen Wirkung auf IL-6 in Zusammenhang (siehe Rubrik «Präklinische Daten, Reproduktionstoxizität»). Die klinische Bedeutung dieser Befunde ist nicht bekannt.
Enspryng darf während einer Schwangerschaft nicht angewendet werden, es sei denn, der potentielle Nutzen für die Mutter überwiegt gegenüber dem potentiellen Risiko für den Fötus. Da Satralizumab die Plazentaschranke passiert, kann bei Kindern von mit Enspryng behandelten Frauen ein erhöhtes Infektionsrisiko bestehen; bei der Verabreichung von Lebendimpfstoffen an diese Kinder ist Vorsicht geboten.
Bei Frauen im gebärfähigen Alter sollte die Anwendung einer geeigneten Verhütungsmethode während der Behandlung und bis zu 5 Monate nach der letzten Enspryng Dosis in Betracht gezogen werden.
Stillzeit
Es ist nicht bekannt, ob Enspryng in der menschlichen Muttermilch ausgeschieden oder nach Aufnahme systemisch absorbiert wird. Da aber IgGs in der menschlichen Muttermilch ausgeschieden werden und präklinisch das Ausscheiden in der Muttermilch nachgewiesen worden ist (siehe Rubrik «Präklinische Daten»), sollten Frauen, die mit Enspryng behandelt werden, nicht stillen. Dabei sollen der Nutzen des Stillens für das Kind und der Nutzen der Therapie für die Mutter gegeneinander abgewogen werden.
Fertilität
Es sind keine klinischen Daten zur Auswirkung von Enspryng auf die Fertilität beim Menschen verfügbar. Tierexperimentelle Studien liessen keine Beeinträchtigung der männlichen oder weiblichen Fertilität erkennen (siehe Rubrik «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Es wurden keine Studien zu den Auswirkungen auf die Fahrtüchtigkeit und die Fähigkeit, Maschinen zu bedienen, durchgeführt. Die verfügbaren Daten lassen jedoch nicht erkennen, dass die Behandlung mit Enspryng die Fahrtüchtigkeit und die Fähigkeit zum Bedienen von Maschinen beeinflusst.
Unerwünschte Wirkungen
Zusammenfassung des Sicherheitsprofils
Die Sicherheit von Enspryng als Monotherapie oder in Kombination mit einer IST wurde anhand von Daten aus zwei randomisierten, multizentrischen, doppelblinden, placebokontrollierten klinischen Phase-III-Studien (BN40898 und BN40900) bewertet, in denen 63 Patienten eine Enspryng-Monotherapie und 41 Patienten eine Therapie mit Enspryng in Kombination mit IST erhielten (siehe Rubrik «Eigenschaften/Wirkungen, Klinische Wirksamkeit»).
Die am häufigsten berichteten unerwünschten Arzneimittelwirkungen (UAWs) waren Kopfschmerz, Arthralgie, Leukopenie und injektionsbedingte Reaktionen.
Die verfügbaren Sicherheitsdaten sind hinsichtlich der Anzahl der mit Enspryng exponierten Patienten als auch der Länge der Exposition limitiert. Es kann sein, dass potenzielle, relativ seltene und möglicherweise schwerwiegende UAW im Studienprogramm unerkannt bleiben. Mögliche Klasseneffekte mit anderen IL-6 Rezeptor Inhibitoren wie z.B. Darmperforationen, opportunistische Infekte einschliesslich Tuberkulose, Hepatitis-B Reaktivierung, Erhöhung des kardiovaskuären Risikos, Aktivierung des Komplementsystems, demyelinisierende Erkrankungen, sowie Malignome sind nicht auszuschliessen (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Tabellarische Zusammenfassung unerwünschter Arzneimittelwirkungen aus klinischen Studien
In Tabelle 1 sind die UAWs zusammengefasst, die im Zusammenhang mit der Anwendung von Enspryng als Monotherapie oder in Kombination mit IST in klinischen Studien berichtet wurden. In beiden klinischen Studien wurden die Patienten in den Enspryng-Gruppen länger behandelt als die Patienten in der Placebo-Gruppe (oder in der Gruppe, in der das Placebo in Kombination mit IST angewendet wurde). UAWs aus klinischen Studien (Tabelle 1) sind nach MedDRA-Systemorganklasse aufgeführt. Die jeweilige Häufigkeitskategorie für jede unerwünschte Arzneimittelwirkung basiert auf der folgenden Konvention: sehr häufig (≥1/10), häufig (≥1/100 bis <1/10), gelegentlich (≥1/1'000 bis <1/100), selten (≥1/10'000 bis <1/1'000), sehr selten (<1/10'000).
Tabelle 1: Zusammenfassung von UAWs bei Patienten, die in klinischen Studien mit Enspryng als Monotherapie oder in Kombination mit einer immunsuppressiven Therapie behandelt wurden
Unerwünschte Wirkungen (MedDRA) | Anzahl Patienten (%) | Häufigkeitskategorie bei Enspryng | |
Enspryng | Placebo1 | ||
Infektionen und parasitäre Erkrankungen | |||
Harnwegsinfekte | 18 (17,3%) | 15 (20,3%) | Sehr häufig |
Infektionen der oberen Atemwege | 20 (19,2%) | 12 (16,2%) | Sehr häufig |
Nasopharyngitis | 19 (18,3%) | 8 (10,8%) | Sehr häufig |
Influenza | 5 (4,8%) | 6 (8,1%) | Häufig |
Erkrankungen des Nervensystems | |||
Kopfschmerzen | 20 (19,2%) | 8 (10,8%) | Sehr häufig |
Migräne | 4 (3,8%) | 0 | Häufig |
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen | |||
Injektionsbedingte Reaktionen | 13 (12,5%) | 7 (9,5%) | Sehr häufig |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | |||
Arthralgie | 14 (13,5%) | 1 (1,4%) | Sehr häufig |
Steifigkeit des Bewegungsapparats | 5 (4,8%) | 0 | Häufig |
Erkrankungen der Haut und des Unterhautgewebes | |||
Hautausschlag | 9 (8,7%) | 3 (4,1%) | Häufig |
Pruritus | 6 (5,8%) | 1 (1,4%) | Häufig |
Psychiatrische Erkrankungen | |||
Insomnie | 6 (5,8%) | 1 (1,4%) | Häufig |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |||
Ödem, peripher | 5 (4,8%) | 0 | Häufig |
Erkrankungen des Blutes und des Lymphsystems | |||
Hypofibrinogenämie | 3 (2,9%) | 0 | Häufig |
Erkrankungen der Atemwege, des Brustraums und Mediastinums | |||
Rhinitis, allergisch | 4 (3,8%) | 0 | Häufig |
Stoffwechsel- und Ernährungsstörungen | |||
Hyperlipidämie | 14 (13,5%) | 9 (12,2%) | Sehr häufig |
Untersuchungen | |||
Leukopenie | 14 (13,5%) | 4 (5,4) | Sehr häufig |
Hyperbilirubinämie | 2 (1,9%) | 0 | Häufig |
Anstieg des Körpergewichts | 5 (4,8%) | 2 (2,7%) | Häufig |
Thrombopenie | 4 (3,8%) | 2 (2,7%) | Häufig |
Erhöhung der Transaminasen | 7 (6,7%) | 6 (8,1%) | Häufig |
1 Placebo oder Placebo in Kombination mit IST
Beschreibung ausgewählter Nebenwirkungen
Injektionsbedingte Reaktionen (IRRs)
Die IRRs, die bei Patienten unter Behandlung mit Enspryng als Monotherapie oder in Kombination mit IST berichtet wurden, waren überwiegend leicht bis mittelschwer und traten meist innerhalb von 24 Stunden nach den Injektionen auf. Die am häufigsten berichteten systemischen Symptome waren Diarrhöe und Kopfschmerz. Die am häufigsten berichteten lokalen Reaktionen an der Injektionsstelle waren Wärmegefühl, Erythem, Juckreiz, Hautausschlag und Schmerzen. Keine der injektionsbedingten Reaktionen erforderte eine Unterbrechung oder ein Absetzen der Dosisgabe.
Infektionen
Um der längeren Behandlungsdauer in der Satralizumabgruppe gegenüber der Placebogruppe Rechnung zu tragen, erfolgen die Angaben zu Infektionen nicht nur in Prozent, sondern zusätzlich in Ereignissen pro 100 PJ (Ereignisse, die bei 100 Patienten nach einem Jahr Behandlung auftreten).
In der Studie mit Enspryng als Monotherapie betrugen die Häufigkeiten von Infektionen bei mit Enspryng behandelten Patienten 99,8 Ereignisse/100 PJ (95%-KI: 82,4; 119,8) bzw. 34/63 Patienten [54,0% (95%-KI: 40,94%; 66,61%)] im Vergleich zu 162,6 Ereignisse/100 PJ (95%-KI: 125,8; 206,9) bzw. 14/32 Patienten [43,8% (95%-KI: 26,36%; 62,34%)] bei den Patienten in der Placebogruppe. Die Häufigkeiten schwerwiegender Infektionen betrugen 5,2 Ereignisse/100 PJ (95%-KI: 1,9; 11,3) bzw. 6/63 Patienten [9,5% (95%-KI: 3,58%; 19,59%)] bei mit Enspryng behandelten Patienten im Vergleich zu 9,9 Ereignissen/100 PJ (95%-KI: 2,7; 25,2) bzw. 3/32 Patienten [9,4% (95%-KI: 1,98%; 25,02%)] bei den Patienten in der Placebogruppe.
Bei Patienten unter Behandlung mit Enspryng in Kombination mit einer IST betrugen die Häufigkeiten von Infektionen 132,5 Ereignisse/100 PJ (95%-KI: 108,2; 160,5) bzw. 28/41 Patienten [68,3% (95%-KI: 51,91%; 81,92%)] im Vergleich zu 149,6 Ereignissen/100 PJ (95%-KI: 120,1; 184,1) bzw. 26/42 Patienten [61,9% (95%-KI: 45,64%; 76,43%)] bei Patienten, die ein Placebo in Kombination mit einer IST erhielten. Die Häufigkeiten schwerwiegender Infektionen betrugen 2,6 Ereignisse/100 PJ (95%-KI: 0,3; 9,2) bzw. 2/41 Patienten [4,9% (95%-KI: 0,60%; 16,53%)] im Vergleich zu 5,0 Ereignissen/100 PJ (95%-KI: 1,0; 14,7) bzw. 3/42 Patienten [7,1% (95%-KI: 1,50%; 19,48%)] bei Patienten, die ein Placebo in Kombination mit einer IST erhielten.
Anstieg des Körpergewichts
Im Zeitraum der doppelverblindeten Behandlung wurde ein Anstieg des Körpergewichts um ≥15% gegenüber dem Ausgangswert bei 4,8% der mit Enspryng behandelten Patienten (Monotherapie oder in Kombination mit einer immunsuppressiven Therapie) bzw. bei 2,7% der Patienten, die ein Placebo (oder ein Placebo in Kombination mit einer immunsuppressiven Therapie) erhielten, beobachtet.
Laboranomalien
Neutrophile
In der doppelblinden Behandlungsphase wurden bei 31,7% der Patienten, die mit Enspryng (als Monotherapie oder in Kombination mit IST) behandelt wurden, und bei 21,6% der Patienten, die das Placebo (allein oder kombiniert mit einer IST) erhielten, verringerte Neutrophilenzahlen gemessen. Die Verringerung der Neutrophilenzahlen war meistens vorübergehend oder intermittierend.
Von den Patienten im Enspryng-Arm hatten 9,6% einen Neutrophilenwert unter 1 x 109/l, verglichen mit 5,4% im Placebo-Arm oder im Placebo-plus-IST-Arm, was zeitlich nicht mit schwerwiegenden Infektionen einherging.
Thrombozyten
In der doppelblinden Behandlungsphase wurden bei 24,0% der Patienten, die mit Enspryng (als Monotherapie oder in Kombination mit IST) behandelt wurden, und bei 9,5% der Patienten, die das Placebo (allein oder kombiniert mit einer IST) erhielten, verringerte Thrombozytenzahlen gemessen. Die verringerten Thrombozytenzahlen waren nicht mit Blutungsereignissen verbunden.
Die Verringerung der Thrombozytenzahlen war vorübergehend und lag nicht unter 75 x 109/l. Bei keinem Patienten fiel der Thrombozytenwert auf ≤50 x 109/l.
Leberenzyme
In der doppelblinden Behandlungsphase wurden bei 27,9% bzw. 18,3% der Patienten, die mit Enspryng (als Monotherapie bzw. in Kombination mit IST) behandelt wurden, und bei 12,2% bzw. 13,5% der Patienten, die das Placebo (allein bzw. kombiniert mit einer IST) erhielten, Erhöhungen der ALT- oder AST-Werte gemessen. Die Erhöhungen lagen meist unter dem 3-Fachen der ONG, waren vorübergehend und ohne Unterbrechung der Enspryng-Gabe reversibel.
ALT- oder AST-Erhöhungen >3 x ONG traten bei 2,9% bzw. 1,9% der mit Enspryng als Monotherapie bzw. in Kombination mit IST behandelten Patienten auf, gingen aber nicht mit einem Anstieg des Gesamtbilirubins einher. Bei einem Patienten, der Enspryng in Kombination mit einer IST erhielt, wurden 4 Wochen nach Therapiebeginn ALT-Erhöhungen >5 x ONG gemessen, die sich nach dem Absetzen von Enspryng normalisierten.
Lipidwerte
In der doppelblinden Behandlungsphase wurden bei 10,6% der Patienten, die mit Enspryng (als Monotherapie oder in Kombination mit IST) behandelt wurden, und bei 1,4% der Patienten, die das Placebo (allein oder kombiniert mit einer IST) erhielten, Erhöhungen des Gesamtcholesterins auf über 7,75 mmol/l gemessen. Bei 18,3% der mit Enspryng behandelten Patienten traten Erhöhungen der Triglyzeride auf über 3,42 mmol/l auf, verglichen mit 6,8% der Patienten bei Placebogabe. Die Erhöhungen der Lipidwerte erforderten keine Dosisunterbrechung.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Es wurden keine Fälle von Überdosierung bei Patienten mit NMO oder NMOSD berichtet. Es liegen keine Erfahrungen mit Überdosierungen bei Patienten mit NMO oder NMOSD vor.
Anzeichen und Symptome
In einer Phase-I-Studie erhielten gesunde erwachsene Probanden subkutan eine Einzeldosis von bis zu 240 mg Enspryng, ohne dass schwerwiegende oder schwere unerwünschte Ereignisse auftraten.
Behandlung
Im Falle einer Überdosierung sollte der Patient engmaschig überwacht und symptomorientiert behandelt werden. Bei Bedarf sind unterstützende Massnahmen einzuleiten.
Eigenschaften/Wirkungen
ATC-Code:
L04AC19
Wirkungsmechanismus
Satralizumab ist ein humanisierter monoklonaler IgG2-Antikörper (mAk), der an löslichen und membrangebundenen humanen IL-6-Rezeptor (IL-6R) bindet und dadurch die IL-6 vermittelte Signalübertragung durch diese Rezeptoren verhindert.
IL-6 ist ein pleiotropes Zytokin, das von einer Vielzahl von Zelltypen gebildet wird und an verschiedenen Entzündungsprozessen beteiligt ist, beispielsweise an der Aktivierung von B-Zellen, der Differenzierung von B-Zellen zu Plasmablasten und bei der Produktion von Autoantikörpern, bei der Aktivierung und Differenzierung von Th17-Zellen, bei der Hemmung regulatorischer T-Zellen und bei Veränderungen der Durchlässigkeit der Blut-Hirn-Schranke. Die IL-6-Spiegel im Liquor und im Serum von Patienten mit NMO und NMOSD sind während Phasen mit aktiver Krankheit erhöht. Einige IL-6-Funktionen werden mit der Pathogenese von NMO und NMOSD in Verbindung gebracht, darunter die Bildung pathologischer Autoantikörper gegen Aquaporin-4 (AQP4), ein Wasserkanalprotein, das hauptsächlich von Astrozyten im ZNS exprimiert wird.
Pharmakodynamik
In klinischen Studien mit Enspryng bei NMO und NMOSD wurden Verringerungen der Werte für das C-reaktive Protein (CRP), Fibrinogen und Komplement (C3, C4 und CH50) festgestellt.
Klinische Wirksamkeit
Die Wirksamkeit und Sicherheit von Enspryng wurden in zwei klinischen Pivotstudien der Phase III (BN40898 und BN40900) bei Patienten mit diagnostizierter AQP4-IgG-seropositiver oder -seronegativer NMO (Kriterien in Wingerchuk 2006) oder mit diagnostizierter AQP4-IgG-seropositiver NMOSD (Kriterium in Wingerchuk 2007) beurteilt. Rückblickend erfüllten diese Patienten auch die neuesten, vom internationalen Gremium für die Diagnose von NMO vorgeschlagenen Kriterien (Ref: Wingerchuk et al. 2015). Die Auswirkungen von Enspryng wurden bei erwachsenen Patienten (in den Studien BN40898 und BN40900) und bei jugendlichen Patienten (im Alter von ≥12 bis <18 Jahren, insgesamt N=7 (N=4 Enspryng, N=3 Placebo) in der Studie BN40898) untersucht. Der Einschluss erwachsener NMO-Patienten, die AQP4-IgG-seronegativ waren, war in beiden Studien auf ungefähr 30% beschränkt, damit das Studienkollektiv das reale NMO-Patientenkollektiv widerspiegelte.
Der primäre Indikator für die Wirksamkeit war in beiden Studien die Zahl der prüfplangemäss definierten Rezidive (PDRs), basierend auf einer vorab festgelegten Verschlechterung der Ergebnisse der EDSS (Expanded Disability Status Scale) und des FSS (Functional System Scores), die von einem unabhängigen CEC (Clinical Endpoint Committee) bestätigt wurden. Der primäre Endpunkt für die Analyse war die Dauer des Zeitraums bis zum ersten, vom CEC bestätigten PDR mit EDSS/FSS-Beurteilung innerhalb von 7 Tagen nach Meldung der Symptome durch den Patienten (adjudiziertes Rezidiv).
Studie BN40898 (SA-307JG bzw. SAkuraSky)
Die Studie BN40898 war eine randomisierte, multizentrische, doppelblinde, placebokontrollierte klinische Studie zur Bewertung der Wirkung von Enspryng in Kombination mit einer stabilen IST (OKs bis zu 15 mg/Tag [Prednisolonäquivalent], AZA bis zu 3 mg/kg/Tag oder MMF bis zu 3'000 mg/Tag; Jugendliche erhielten eine Kombination aus AZA und OKs oder MMF und OKs). An der Studie nahmen 83 AQP4-IgG-seropositive und -seronegative Patienten (darunter 7 Jugendliche) teil. Die Patienten erhielten die ersten 3 Einzeldosen mit 120 mg Enspryng oder ein entsprechendes Placebo per subkutaner Injektion in die Bauchdecke oder den Oberschenkel alle 2 Wochen während der ersten 4 Wochen und danach einmal alle 4 Wochen.
Das Studiendesign und die Ausgangscharakteristika des Studienkollektivs sind in Tabelle 2 dargestellt.
Die Studie war ereignisgesteuert und die doppelblinde Studienphase zur Bewertung der Wirksamkeit endete, nachdem 26 adjudizierte Rezidive aufgetreten waren. Patienten, bei denen während der doppelverblindeten (DB) Phase ein vom CEC bestätigtes PDR auftrat, die aufgrund eines Rezidivs eine Rescue-Therapie erhielten oder die die DB-Phase abschlossen, konnten in die offene Erweiterungsphase (open-label extension – OLE) aufgenommen werden, bei der alle Patienten unverblindet mit Enspryng behandelt wurden.
Tabelle 2: Studiendesign und Ausgangscharakteristika der Studie BN40898
Studienname | Studie BN40898 (N=83) | |
Studiendesign | ||
Studienpopulation | Jugendliche und erwachsene Patienten mit NMO oder NMOSD mit stabiler Behandlung mit einer IST Alter 12-74 Jahre, ≥2 Rezidive in den letzten 2 Jahren vor dem Screening (mit mindestens einem Rezidiv in den 12 Monaten vor dem Screening), EDSS 0 bis 6,5 | |
Studiendauer zur Bewertung der Wirksamkeit | Ereignisgesteuert (26 CEC-bestätigte, per Prüfplan definierte Rezidive) Mediane Dauer der Nachbeobachtung: Enspryng 100 Wochen, Placebo 74 Wochen | |
Behandlungsgruppen, 1:1 Randomisierung | Gruppe A: 120 mg Enspryng s. c. Gruppe B: Placebo | |
Ausgangscharakteristika | Enspryng + IST (n = 41) | Placebo + IST (n = 42) |
Diagnose, n (%): |
|
|
NMO NMOSD | 33 (80,5) 8 (19,5) | 28 (66,7) 14 (33,3) |
AQP4-IgG-seropositiver Status, n (%) | 27 (65,9) | 28 (66,7) |
Mittleres Alter in Jahren (Standard-Abweichung) | 40,8 (16,1) (13 – 73) | 43,4 (12,0) (14 – 65) |
Jugendliche (≥12 bis <18 Jahre), n (%) | 4 (9,8) | 3 (7,1) |
Geschlechterverteilung, |
|
|
n (%) männlich/ n (%) weiblich | 4 (9,8) / 37 (90,2) | 2 (4,8) / 40 (95,2) |
Immunsuppressive Therapie (IST), n (%): |
|
|
Orale Kortikosteroide (OKs) Azathioprin (AZA) Mycophenolatmofetil (MMF) AZA + OKs* MMF + OKs* | 17 (41,5) 16 (39,0) 4 (9,8) 3 (7,3) 1 (2,4) | 20 (47,6) 13 (31,0) 8 (19,0) 0 1 (2,4) |
* Kombination für jugendliche Patienten zulässig
Studie BN40900 (SA-309JG bzw. SAkuraStar)
Die Studie BN40900 war eine randomisierte, multizentrische, doppelblinde, placebokontrollierte klinische Studie zur Bewertung der Wirkung einer Enspryng-Monotherapie im Vergleich zu einem Placebo. An der Studie nahmen 95 AQP4-IgG-seropositive und -seronegative erwachsene Patienten teil. Die Patienten erhielten die ersten 3 Einzeldosen mit 120 mg Enspryng oder ein entsprechendes Placebo per subkutane Injektion in die Bauchdecke oder den Oberschenkel alle 2 Wochen während der ersten 4 Wochen und danach einmal alle 4 Wochen.
Das Studiendesign und die Ausgangscharakteristika der Studienpopulation sind in Tabelle 3 dargestellt.
Die doppelblinde Studienphase zur Bewertung der Wirksamkeit endete 1,5 Jahre nach dem Datum der Randomisierung des letzten aufgenommenen Patienten. Patienten, bei denen während der DB-Phase ein vom CEC bestätigtes PDR auftrat oder die die DB-Phase abschlossen, konnten in die OLE-Phase aufgenommen werden, bei der alle Patienten unverblindet mit Enspryng behandelt wurden.
Tabelle 3: Studiendesign und Ausgangs-Charakteristika der Studie BN40900
Studienname | Studie BN40900 (N = 95) | |
Studiendesign | ||
Studienpopulation | Erwachsene Patienten mit NMO oder NMOSD Alter 18-74 Jahre, ≥1 Rezidiv oder erste Attacke in den letzten 12 Monaten vor dem Screening, EDSS 0 bis 6,5. Die Patienten hatten entweder eine Vorbehandlung zur Verhinderung eines NMOSD-Rezidivs erhalten oder waren unbehandelt. | |
Studiendauer zur Bewertung der Wirksamkeit | Ereignisgesteuert (44 CEC-bestätigte, per Prüfplan definierte Rezidive oder 1,5 Jahre nach dem Datum der Randomisierung des letzten aufgenommenen Patienten, je nachdem, was zuerst eintritt) Mediane Dauer der Nachbeobachtung: Enspryng 95,4 Wochen, Placebo 60,5 Wochen | |
Behandlungsgruppen, 2:1 Randomisierung | Monotherapie: Gruppe A: 120 mg Enspryng s. c. Gruppe B: Placebo | |
Ausgangscharakteristika | Enspryng (n = 63) | Placebo (n = 32) |
Diagnose, n (%): |
|
|
NMO NMOSD | 47 (74,6) 16 (25,4) | 24 (75,0) 8 (25,0) |
AQP4-IgG-seropositiver Status, n (%) | 41 (65,1) | 23 (71,9) |
Mittleres Alter in Jahren (Standard-Abweichung) | 45,3 (12,0) (21 – 70) | 40,5 (10,5) (20 – 56) |
Geschlechterverteilung, |
|
|
n (%) männlich/ n (%) weiblich | 17 (27,0) / 46 (73,0) | 1 (3,1) / 31 (96,9) |
Primäre Wirksamkeit - doppelverblindete Phase
In der ITT Population führte die Behandlung mit Enspryng zu einer statistisch signifikanten Verringerung des Risikos des Auftretens eines adjudizierten Rezidivs um 62% (Hazard Ratio [HR] [95%-KI]: 0,38 [0,16-0,88]; p [Log-Rang-Test] = 0,0184) bei Anwendung in Kombination mit einer stabilen IST (Studie BN40898), und bei Anwendung als Monotherapie ergab sich eine Verringerung des Risikos des Auftretens eines adjudizierten Rezidivs um 55% (HR [95%-KI]: 0,45 [0,23-0,89]; p [Log-Rang-Test] = 0,0184) (Studie BN40900), jeweils im Vergleich zum Placebo.
Der stärkste Teilgruppeneffekt wurde bei den AQP4-IgG seropositiven Patienten beobachtet. Bei AQP4-IgG-seropositiven Patienten war das relative Risiko des Auftretens eines adjudizierten Rezidivs in Studie BN40898 um 79% reduziert (HR [95%-KI]: 0,21 [0,06-0,75]) und in Studie BN40900 um 74% (HR [95%-KI]: 0,26 [0,11-0,63]). Nach 48 Wochen waren 91,5% und 82,9% der mit Enspryng in Kombination mit einer IST bzw. als Monotherapie behandelten AQP4-IgG-seropositiven Patienten adjudiziert rezidivfrei geblieben. Nach 96 Wochen waren 91,5% und 76,5% der mit Enspryng in Kombination mit einer IST bzw. als Monotherapie behandelten AQP4-IgG-seropositiven Patienten adjudiziert rezidivfrei geblieben (siehe Tabelle 4 und Abb. 1 und 2). Wurden Daten aus den Studien BN40898 und BN40900 kombiniert, führte die Behandlung mit Enspryng mit oder ohne IST bei AQP4-IgG-seropositiven Patienten zu einer Verringerung des Risikos um insgesamt 75% (HR [95%-KI]; 0,25 [0,12-0,50]). Die Unterschiede hinsichtlich der Dauer des Zeitraums bis zum ersten adjudizierten Rezidiv in der Gruppe der AQP4-IgG-seronegativen Patienten zwischen solchen, die Enspryng mit oder ohne IST erhielten, und solchen, die ein Placebo mit oder ohne IST erhielten, waren nicht signifikant (BN40898 und BN40900 kombiniert: HR [95%-KI]: 0,97 [0,41-2,33]).
Tabelle 4: Wichtigste Wirksamkeitsendpunkte in den Studien BN40898 und BN40900 bei AQP4-IgG-seropositiven Patienten
BN40898 | BN40900 | |||
Enspryng | Placebo | Enspryng | Placebo | |
Primärer Endpunkt bei AQP4-IgG-seropositiven Patienten | ||||
Anzahl der AQP4-IgG-seropositiven Patienten (n) | 27 | 28 | 41 | 23 |
Risiko Verminderung (Individuelle Studien) | 79% (HR: 0,21; 95%-KI: 0,06; 0,75; p = 0,0086) | 74% (HR: 0,26; 95%-KI: 0,11; 0,63; p = 0,0014) | ||
Risiko Verminderung (gepoolte Analyse) | 75% (HR: 0,25; 95%-KI: 0,12; 0,50; p: <0,0001) | |||
Anteil der adjudizierten rezidivfreien Patienten nach 48 Wochen | 91,5% (95%-KI: 69,64; 97,83) | 59,9% (95%-KI: 36,25; 77,25) | 82,9% (95%-KI: 67,49; 91,47) | 55,4% (95%-KI: 32,96; 73,08) |
Anteil der adjudizierten rezidivfreien Patienten nach 96 Wochen | 91,5% (95%-KI: 69,64; 97,83) | 53,3% (95%-KI: 29,34; 72,38) | 76,5% (95%-KI: 59,22; 87,21) | 41,1% (95%-KI: 20,76; 60,41) |
Abb. 1: Studie BN40898: Dauer des Zeitraums bis zum ersten adjudizierten Rezidiv während der Doppelblindphase bei AQP4-IgG-seropositiven Patient

Abb. 2: Studie BN40900: Dauer des Zeitraums bis zum ersten adjudizierten Rezidiv während der Doppelblindphase bei AQP4-IgG-seropositiven Patienten

Die Behandlung AQP4-IgG-seropositiver Patienten mit Enspryng verringerte die annualisierte Rate der adjudizierten Rezidive (Adjudicated Relapse Rate – ARR) im Vergleich zum Placebo in der Studie BN40898 um 88% und in der Studie BN40900 um 90% (Tabelle 5).
Tabelle 5: Annualisierte Rate der adjudizierten Rezidive während der doppelverblindeten Phase (Negativ-Binomial-Regressionsmodel)
BN40898 | BN40900 | Gepoolt | |||||
Placebo | Enspryng | Placebo | Enspryng | Placebo | Enspryng | ||
AQP4-IgG seropositive Patienten | N = 28 | N = 27 | N = 23 | N = 41 | N = 51 | N = 68 | |
Patienten mit Rezidiv | 12 | 3 | 13 | 9 | 25 | 12 | |
Angepasste annualisierte Rezidivrate | 0,520 | 0,063 | 2,853 | 0,275 | 1,339 | 0,136 | |
Relative ARR-Verringerung (Ratenverhältnis) | 88% (RR: 0,122; 95% KI: 0,027; 0,546; p=0,0039) | 90% (RR: 0,096; 95% KI: 0,020; 0,473; p= 0,0086) | 90% (RR: 0,102; 95% KI: 0,034; 0,301; p=0,0002) | ||||
Im Vergleich zu den Patienten, die mit Placebo behandelt wurden, benötigten mit Enspryng behandelte, AQP4-IgG-seropositive Patienten seltener eine Rescue-Therapie (etwa Kortikosteroide, intravenöse Immunglobuline und/oder Apherese [einschl. Plasmapherese und Plasmaaustausch]), und zwar um 61% in der Studie BN40898 und um 74% in der Studie BN40900.
Tabelle 6: Rückgriff auf eine Rescue-Therapie bei Patienten mit Rezidiv während der doppelverblindeten Phase
BN40898 | BN40900 | Gepoolt | ||||
Placebo | Enspryng | Placebo | Enspryng | Placebo | Enspryng | |
AQP4-IgG seropositive Patienten | N = 28 | N = 27 | N = 23 | N = 41 | N = 51 | N = 68 |
Patienten mit Rescue-Therapie | 18 (64,29%) | 11 (40,74%) | 14 (60,87%) | 13 (31,71%) | 32 (62.75%) | 24 (35.29%) |
Risikoreduktion (Odds Ratio) | 61% (OR: 0,3930; 95% KI: 0,1343; 1,1502 p=0,0883) | 74% (OR: 0,2617; 95% KI: 0,0862; 0,7943; p=0,0180) | 66% (OR: 0,3430; 95% KI: 0,1614; 0,7289; p=0,0054) | |||
Die Behandlung AQP4-IgG-seropositiver Patienten mit Enspryng verringerte im Vergleich zum Placebo das Risiko eines schweren Rezidivs (definiert als EDSS-Anstieg um ≥2 Punkte gegenüber der vorgängigen EDSS-Bewertung) in der Studie BN40898 um 85% und in der Studie BN40900 um 79% (Tabelle 7).
Tabelle 7: Zeit bis zum ersten adjudizierten Rezidiv während der doppelverblindeten Phase
BN40898 | BN40900 | Gepoolt | ||||
Placebo | Enspryng | Placebo | Enspryng | Placebo | Enspryng | |
AQP4-IgG seropositive Patienten | N=27 | N=27 | N=23 | N=41 | N=50 | N=68 |
Patienten mit Ereignis | 6 (22,2%) | 1 (3,7%) | 5 (21,7%) | 3 (7,3%) | 11 (22,0%) | 4 (5,9%) |
Risikoreduktion | 85% (HR: 0,15; 95% KI: 0,02; 1,25; p=0,0441) | 79% (HR: 0,21; 95% KI: 0,05; 0,91; p=0,0231) | 82% (HR: 0,18; 95% KI: 0,06; 0,58; p=0,0015) | |||
Die Behandlung von AQP4-IgG-seropositiven Patienten mit Enspryng verringerte das Risiko einer Verschlechterung auf der EDSS gegenüber dem Ausgangswert in der Studie BN40898 um 65% und in der Studie BN40900 um 66% verglichen mit der Placebobehandlung (Tabelle 8).
Tabelle 8: Zeit bis zur Verschlechterung der Scores auf der erweiterten Skala zur Beurteilung des Behinderungsstatus (Expanded Disability Status Scale, EDSS)* in der doppelblinden Phase (AQP4-IgG-seropositive Patienten)
BN40898 | BN40900 | Kombiniert | |||||
Placebo | Enspryng | Placebo | Enspryng | Placebo | Enspryng | ||
AQP4-IgG-seropositive Patienten | N = 27 | N = 28 | N = 23 | N = 41 | N = 50 | N = 69 | |
Patienten mit einem Ereignis | 12 (44,4%) | 5 (17,9%) | 11 (47,8%) | 11 (26,8%) | 23 (46,0%) | 16 (23,2%) | |
Risikoreduzierung | 65% (HR: 0,35; 95%-KI: 0,12; 1,00; p = 0,0407) | 66% (HR: 0,34; 95%-KI: 0,14; 0,82; p = 0,0124) | 69% (HR: 0,31; 95%-KI: 0,16; 0,59; p = 0,0002) | ||||
* Eine Verschlechterung auf der EDSS gegenüber dem Ausgangswert war definiert als: a) Verschlechterung des EDSS-Scores um ≥2 Punkte bei Patienten mit einem Anfangsscore von 0, b) Verschlechterung des EDSS-Scores um ≥1 Punkt(e) bei Patienten mit einem Anfangsscore von 1 bis 5, c) Verschlechterung des EDSS-Scores um ≥0,5 Punkte bei Patienten mit einem Anfangsscore von ≥5,5.
Offene Erweiterungsphase
Analysen langfristiger Daten, darunter jener aus der laufenden offenen Erweiterungsphase (beruhend auf Rezidiven, die mit einer Rescue-Therapie behandelt wurden), ergaben, dass 58% bzw. 73% der mit Enspryng behandelten, AQP4-IgG-seropositiven Patienten nach 120-wöchiger Behandlung rezidivfrei blieben, wenn Enspryng als Add-on- oder als Monotherapie verabreicht wurde.
Insgesamt liegen für die Behandlung mit Enspryng über 2 Jahre hinaus nur limitierte Daten zu Wirksamkeit und Sicherheit vor.
Abb. 3: Studie BN40898: Zeit bis zum ersten Rezidiv (behandeltes klinisches Rezidiv) während der doppelverblindeten und offenen Phase bei AQP4-IgG-seropositiven Patienten

Abb. 4: Studie BN40900: Zeit bis zum ersten Rezidiv (behandeltes klinisches Rezidiv) während der doppelverblindeten und offenen Phase bei AQP4-IgG-seropositiven Patienten

Charakteristika zum Baseline-Zeitpunkt und Wirksamkeit bei jugendlichen Patienten (Studie BN40898)
Das mittlere Alter der 7 jugendlichen Patienten, die während der doppelblinden Phase in die Studie BN40898 aufgenommen wurden, betrug 15,4 Jahre und das mediane Körpergewicht betrug 79,6 kg. Die meisten dieser jugendlichen Patienten waren weiblich (n = 6). Vier Patienten waren weiss, 2 Patienten waren schwarz/Afroamerikaner und 1 Patient war asiatisch. Drei der 7 (42,9%) jugendlichen Patienten waren beim Screening AQP4-IgG-seropositiv (2 in der Placebogruppe und 1 in der Enspryng-Gruppe). In der Doppelblindphase trat bei 1 von 3 Jugendlichen in der Placebo-Gruppe und bei 1 von 4 Jugendlichen in der Enspryng-Gruppe ein adjudiziertes Rezidiv auf. Aufgrund der kleinen Fallzahl wurde die Hazard Ratio für den primären Endpunkt, die Dauer des Zeitraums bis zum ersten adjudizierten Rezidiv, in dieser Teilgruppe nicht berechnet.
Weitere Informationen
Immunogenität
In der Phase-III-Studie BN40898 (Kombination mit IST) und in der Phase-III-Studie BN40900 (Monotherapie) wurden bei 41% bzw. 71% der Patienten, die in der Doppelblindphase Enspryng erhielten, Anti-Arzneistoff-Antikörper (ADAs) festgestellt. Inwieweit diese ADAs fähig sind, die Enspryng-Bindung zu neutralisieren, ist nicht bekannt.
Die Exposition war bei ADA-positiven Patienten geringer, es gab jedoch keine Auswirkungen der ADAs auf die Sicherheit und keine Auswirkungen wurden auf die Wirksamkeit oder pharmakodynamische Marker, die auf eine Bindung an Zielmoleküle hindeuten, festgestellt.
Die Behandlung mit Satralizumab führte in den Phase-III-Studien zu einer ähnlichen Verringerung des Risikos des Auftretens eines adjudizierten Rezidivs bei den Patienten, trotz jeweils unterschiedlicher Häufigkeiten des Vorhandenseins von ADAs. Patienten mit höherem Körpergewicht und geringerer Exposition entwickelten mit höherer Wahrscheinlichkeit ADAs (unabhängig von der Basisbehandlung mit IST), der Behandlungseffekt war jedoch in allen Körpergewichtsgruppen vergleichbar, wenn die Anwendung entweder in Kombination mit IST oder als Monotherapie erfolgte.
Die empfohlene Dosis ist für alle Patienten geeignet und bei Patienten, die ADAs entwickeln, ist weder eine Unterbrechung noch eine Änderung der Dosis erforderlich.
Pharmakokinetik
Die Pharmakokinetik von Enspryng wurde bei japanischen und kaukasischen gesunden Probanden und bei Patienten mit NMO und NMOSD beschrieben. Die Charakterisierung der Pharmakokinetik bei Patienten mit NMO und NMOSD bei Anwendung der empfohlenen Dosis erfolgte anhand von Populationspharmakokinetischen Analysen.
Der zeitabhängige Verlauf der Konzentration von Enspryng bei Patienten mit NMO oder NMOSD konnte durch ein Zweikompartiment-Populations-PK-Modell mit paralleler linearer und zielvermittelter (Michaelis-Menten-Kinetik) Elimination und subkutaner (SC)-Absorption erster Ordnung beschrieben werden. Enspryng-Clearance- und -Volumenparameter wurden allometrisch nach Körpergewicht skaliert (anhand einer Power-Funktion mit dem festen Power-Koeffizienten von 0,75 bzw. 1 für Clearance- bzw. Volumenparameter). Das Körpergewicht erwies sich als signifikante Kovariante, wobei Clearance und Vc (zentrales Verteilungsvolumen) bei Patienten mit einem Gewicht von 123 kg (97,5tes Perzentil der Gewichtsverteilung) im Vergleich zu einem 60 kg schweren Patienten um 71,3% bzw. 105% anstiegen.
Die Steady-State-Pharmakokinetik wurde nach der Aufsättigungsphase (8 Wochen) mit folgenden Werten für Cmin, Cmax und AUC (Mittelwert (± Std.-Abw.)) erreicht: Cmin: 28,5 (10,2) µg/ml, Cmax: 42,7 (11,9) µg/ml und AUC: 1020 (313) µg.ml/Tag. Die Pharmakokinetik wurde durch die Basisimmuntherapie nicht beeinflusst (siehe Rubrik «Interaktionen»).
Absorption
Der Absorptionsgeschwindigkeitskonstante von Enspryng betrug 0,251 1/Tag (95%-KI: 0,216-0,285), was einer Absorptionshalbwertszeit von ungefähr 3 Tagen bei Anwendung der empfohlenen Dosis entspricht (siehe Rubrik «Dosierung/Anwendung»). Die Bioverfügbarkeit war hoch (85,4%, 95%-KI: 0,795-0,953).
Distribution
Enspryng unterliegt einer biphasischen Distribution. Das zentrale Distributionsvolumen betrug 3,46 l (95%-KI: 3,21-3,97), das periphere Distributionsvolumen 2,07 l (95%-KI: 1,78-2,59). Die interkompartmentale Clearance betrug 0,336 l/Tag (95%-KI: 0,261-0,443).
Metabolismus
Der Metabolismus von Enspryng wurde nicht direkt untersucht. Es wird angenommen, dass Enspryng, katabolisch eliminiert wird.
Elimination
Die Clearance von Enspryng ist konzentrationsabhängig. Die lineare Clearance beträgt schätzungsweise 0,0601 l/Tag (95%-KI: 0,0524-0,0695) und macht etwa die Hälfte der Gesamtclearance im Steady-State bei Anwendung der empfohlenen Dosis bei Patienten mit NMO und NMOSD aus. Auf der Basis von kombinierten Daten aus den Phase-3-Studien beträgt die damit zusammenhängende terminale t1/2 ungefähr 30 Tage (Bereich 22-37 Tage).
Kinetik spezieller Patientengruppen
In Analysen der Populationspharmakokinetik bei erwachsenen Patienten mit NMO oder NMOSD wurden keine Effekte des Alters, Geschlechts und der ethnischen Zugehörigkeit auf die Pharmakokinetik von Satralizumab gefunden. Wenngleich das Körpergewicht Einfluss auf die Pharmakokinetik von Satralizumab hatte, bestehen keine Empfehlungen zur Anpassung der Dosis, um diesen demografischen Merkmalen Rechnung zu tragen.
Leberfunktionsstörungen
Es wurde keine formale Studie zur Auswirkung einer Leberfunktionsstörung auf die PK von Satralizumab durchgeführt.
Nierenfunktionsstörungen
Es wurde keine formale Studie zur Auswirkung einer Nierenfunktionsstörung auf die PK von Satralizumab durchgeführt. An den klinischen Studien BN40898 und BN40900 nahmen jedoch 22 Patienten mit leichter Nierenfunktionsstörung (Kreatinin-Clearance <80 ml/min und ≥50 ml/min) teil. Wie aufgrund der bekannten Clearance-Mechanismen für Satralizumab zu erwarten war, war die PK bei diesen Patienten nicht beeinträchtigt, und daher ist keine Dosisanpassung erforderlich.
Ältere Patienten
Es wurden keine speziellen Studien durchgeführt, um die PK von Satralizumab bei Patienten über 65 Jahren zu untersuchen. An den klinischen Studien BN40898 und BN40900 nahmen jedoch Patienten mit NMO oder NMOSD im Alter zwischen 65 und 74 Jahren teil.
Analysen der Populationspharmakokinetik basierend auf Daten dieser Patienten zeigten, dass das Alter keinen Einfluss auf die PK von Satralizumab hatte.
Kinder und Jugendliche
Daten von 8 jugendlichen Patienten [13-17 Jahre], bei denen das Dosierungsregime für Erwachsene angewendet worden war, zeigen, dass sich die Populations-PK-Parameter von Satralizumab nicht signifikant von denen bei Erwachsenen unterscheiden.
Es ist daher keine Dosisanpassung erforderlich.
Präklinische Daten
Sicherheitspharmakologie/Langzeittoxizität (bzw. Toxizität bei wiederholter Verabreichung)
Präklinische Studien an Affen, einer Responder-Spezies mit Kreuzreaktivität gegenüber Satralizumab, liessen basierend auf der Sicherheitspharmakologie und auf Toxizitätsendpunkten keine besonderen Gefährdungen für den Menschen erkennen. Bei einmal wöchentlicher wiederholter subkutaner Verabreichung von bis zu 50 mg Satralizumab pro kg an Cynomolgus-Affen in 4- und 26-wöchigen Toxizitätsstudien wurden keine adversen Effekte beobachtet. Die einzige relevante Änderung in diesen Studien war ein Anstieg des IL-6-Spiegels im Blut, der als Ergebnis der pharmakologischen Wirkung (Blockade des IL-6R) von Satralizumab betrachtet wurde. Die Behandlung mit Satralizumab löste bei den meisten behandelten Tieren eine Immunantwort mit Anti-Arzneistoff-Antikörpern aus, was jedoch die pharmakologische Antwort nicht beeinflusste und nicht zu unerwünschten Ereignissen führte.
Gentoxizität
Es wurden keine Studien zur Feststellung des gentoxischen Potenzials von Satralizumab durchgeführt. Es ist nicht davon auszugehen, dass Antikörper Auswirkungen auf die DNA haben.
Kanzerogenität
Es wurden keine Kanzerogenitätsstudien mit Satralizumab durchgeführt. In einer 6-monatigen Studie an Cynomolgus-Affen zur chronischen Toxizität wurden keine proliferativen Läsionen festgestellt.
Fertilität
Bei Affen hatte die Langzeitgabe von Satralizumab keine Auswirkungen auf männliche oder weibliche Fortpflanzungsorgane.
Reproduktionstoxizität
Die Behandlung von trächtigen Cynomolgus-Affen mit bis zu 50 mg Satralizumab pro kg pro Woche bis zur Geburt bewirkte keine nachteiligen Auswirkungen auf die Muttertiere, die Entwicklung des Fötus, den Ausgang der Trächtigkeit oder das Überleben und die Entwicklung der Jungtiere, einschliesslich der Lernfähigkeit. Die Antikörperreaktion auf ein T-Zell-abhängiges Antigen war in Nachkommen von Satralizumab-behandelten Tieren niedriger als in Nachkommen von Kontrolltieren, was wahrscheinlich in Zusammenhang mit der pharmakologischen Wirkung auf IL-6 steht.
Die Nachkommen Satralizumab-behandelter Muttertiere wiesen eine systemische Exposition auf und zeigten bis zu 6 Monate nach Geburt erhöhte IL-6-Spiegel im Plasma. Satralizumab wurde in der Milch nachgewiesen (<0,9% der jeweiligen Plasmaspiegel des Muttertiers).
Sonstige Hinweise
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8 °C) lagern.
Ungeöffnet kann Enspryng aus dem Kühlschrank entnommen und wieder darin zurückgestellt werden, falls dies erforderlich ist. Bei Aufbewahrung bei Raumtemperatur ohne Kühlung (max. 30 °C) sollte eine Dauer von 8 Tagen nicht überschritten werden.
Nicht schütteln. Nicht einfrieren.
Den Behälter im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Enspryng ist nur für die Einmalanwendung bestimmt.
Die Fertigspritze vor der Anwendung 30 Minuten ausserhalb der Faltschachtel bei Raumtemperatur aufbewahren.
Das Arzneimittel nicht injizieren, wenn die Flüssigkeit trüb oder verfärbt ist oder Partikel enthält.
Die Fertigspritze und den Nadelschutz auf Beschädigungen überprüfen. Bei Vorhandensein von Rissen oder Defekten nicht verwenden.
Entsorgung von Fertigspritze und Nadelschutz
Folgende Punkte sind bei der Verwendung und Entsorgung der Fertigspritze und des Nadelschutzes unbedingt zu beachten:
- Die Fertigspritze darf auf keinen Fall wiederverwendet werden.
- Die benutzte Spritze sofort nach Gebrauch in einen Entsorgungsbehälter für scharfe und spitze Gegenstände geben.
- Die Fertigspritze und den Nadelschutz nach den vor Ort geltenden Bestimmungen oder den Anweisungen des medizinischen Fachpersonals entsorgen.
- Die Fertigspritze und den Nadelschutz und alle Arzneimittel ausser Reichweite von Kindern aufbewahren.
Entsorgung von nicht verwendeten/abgelaufenen Medikamenten
Die Freisetzung von Arzneimitteln in die Umwelt sollte minimiert werden. Arzneimittel sollten nicht über das Abwasser entsorgt werden. Eine Entsorgung mit dem Haushaltsabfall sollte vermieden werden. Verwenden Sie - falls an Ihrem Standort möglich - etablierte «Sammelsysteme».
Zulassungsnummer
67617 (Swissmedic).
Zulassungsinhaberin
Roche Pharma (Schweiz) AG, Basel.
Stand der Information
Juli 2020.
▼ Questo medicamento è soggetto a monitoraggio addizionale. Ciò consente una rapida identificazione delle nuove conoscenze in materia di sicurezza. Gli appartenenti alle professioni sanitarie sono invitati a segnalare il sospetto di effetti collaterali nuovi o gravi. Per indicazioni a proposito della segnalazione di effetti collaterali, cfr. la rubrica «Effetti indesiderati».
Composizione
Principi attivi
Satralizumabum (prodotto con tecnologia genetica utilizzando cellule CHO [Chinese Hamster Ovary]).
Sostanze ausiliarie
L-histidinum; acidum L-asparticum; L-arginini hydrochloridum; poloxamera 188; aqua ad iniectabilia q.s. ad solutionem pro 1,0 ml.
Forma farmaceutica e quantità di principio attivo per unità
Soluzione iniettabile in una siringa preriempita.
Soluzione sterile pronta all'uso per iniezione sottocutanea (s.c.) in siringa preriempita con dispositivo di protezione dell'ago (Needle Safety Device, NSD), contenente una dose singola.
Enspryng soluzione per iniezione s.c. è un liquido da incolore a giallo chiaro in una siringa preriempita contenente 1 ml di soluzione. Ogni siringa preriempita contiene 120 mg di satralizumab.
Indicazioni/Possibilità d'impiego
Enspryng viene usato in monoterapia o in associazione con una terapia immunosoppressiva per il trattamento delle malattie dello spettro della neuromielite ottica (neuromyelitis optica spectrum disorders, NMOSD) in adulti e adolescenti nei quali sono rilevabili anticorpi IgG anti-aquaporina 4 (che sono cioè sieropositivi alle IgG AQP4).
Posologia/Impiego
Generali
Per garantire la tracciabilità dei medicamenti biotecnologici, si raccomanda di prendere nota del nome commerciale e del numero di lotto in occasione di ogni trattamento.
Dosaggio raccomandato
Enspryng deve essere somministrato con iniezione sottocutanea.
Inizio della terapia/dose di saturazione
La dose di saturazione raccomandata è di 120 mg per iniezione s.c. ogni 2 settimane (prima dose nella settimana 0, seconda dose nella settimana 2 e terza dose nella settimana 4) per le prime tre applicazioni.
Terapia di mantenimento
La dose di mantenimento raccomandata è di 120 mg tramite iniezione s.c. ogni 4 settimane (a partire dalla settimana 8).
Durata della terapia
Enspryng è destinato alla terapia di lunga durata.
Aggiustamento della dose a causa di effetti indesiderati/interazioni
Alterazioni degli enzimi epatici
Il trattamento con Enspryng deve essere interrotto definitivamente quando l'aumento dell'alanina-aminotransferasi (ALT) o dell'aspartato-transaminasi (AST) risulta superiore a cinque volte il limite superiore della norma (LSN) e si accompagna a un aumento della bilirubina.
Se l'aumento dei valori dell'ALT o dell'AST risulta superiore a 5 volte il limite superiore della norma (LSN), ma non si accompagna a un aumento della bilirubina, il trattamento con Enspryng va interrotto. Il trattamento può proseguire (120 mg s.c. ogni 4 settimane) quando i valori di ALT e AST si collocano nuovamente nell'ambito della normalità, anche sulla base di una valutazione del rapporto tra rischio e beneficio del trattamento per il paziente. Quando si decide di proseguire il trattamento, i valori degli enzimi epatici vanno strettamente monitorati. Se in seguito si constata un nuovo aumento di AST/ALT e/o della bilirubina, il medicamento deve essere definitivamente interrotto.
Neutropenia
Se il numero dei neutrofili risulta inferiore a 1,0 x 109/l, ed è confermato in diversi test, il trattamento con Enspryng va interrotto, fino a quando il numero dei neutrofili non sia tornato a valori superiori a 1,0 x 109/l.
Terapia combinata
Enspryng può essere impiegato come monoterapia o in associazione con corticosteroidi per via orale (CO), azatioprina (AZA) o micofenolato mofetile (MMF) (cfr. rubrica «Proprietà/effetti - Efficacia clinica»). Occorre inoltre tener presenti le informazioni professionali di questi preparati.
Istruzioni posologiche speciali
Pazienti con disturbi della funzionalità epatica
La sicurezza e l'efficacia di Enspryng non sono state studiate in pazienti con disturbi della funzionalità epatica (cfr. rubrica «Farmacocinetica - Cinetica di gruppi di pazienti speciali»).
Pazienti con disturbi della funzionalità renale
La sicurezza e l'efficacia di Enspryng non sono state studiate formalmente in pazienti con disturbi della funzionalità renale. Poiché tuttavia Enspryng è un anticorpo monoclonale, che nell'organismo va incontro a processi di degradazione (e non viene rimosso mediante escrezione renale), probabilmente nei pazienti con insufficienza renale non occorre un aggiustamento posologico (cfr. rubrica «Farmacocinetica - Cinetica di gruppi di pazienti speciali»). Negli studi clinici sono stati inclusi pazienti con lieve disfunzione renale, e in questi pazienti la farmacocinetica di satralizumab non risultava modificata (cfr. rubrica «Farmacocinetica - Cinetica di gruppi di pazienti speciali»).
Pazienti anziani
La sicurezza e l'efficacia di Enspryng non sono state studiate in pazienti anziani di età fino ai 74 anni. Nei pazienti di età pari o superiore ai 65 anni non sono necessari aggiustamenti posologici (cfr. rubrica «Farmacocinetica - Cinetica di gruppi di pazienti speciali»).
La sicurezza e l'efficacia di Enspryng non sono state studiate in pazienti di età superiore ai 74 anni (cfr. rubrica «Posologia/impiego - Istruzioni posologiche speciali»).
Bambini e adolescenti
La sicurezza e l'efficacia di Enspryng sono state studiate in un numero limitato (N=4) di pazienti adolescenti di età pari o superiore ai 12 anni. I risultati su farmacocinetica, efficacia e sicurezza erano equiparabili ai risultati ottenuti negli adulti (cfr. rubrica «Proprietà/effetti - Efficacia clinica» e rubrica «Farmacocinetica - Cinetica di gruppi di pazienti speciali»).
La sicurezza e l'efficacia di Enspryng non sono state studiate in pazienti di età inferiore ai 12 anni.
Somministrazione ritardata o saltata della dose
Se si dimentica un'iniezione, la somministrazione deve riprendere non appena possibile e non attendere il momento della successiva dose programmata. Dopo la somministrazione della dose ritardata o saltata, si consiglia di mantenere tra le dosi l'intervallo di somministrazione, rispettivamente di 2 settimane (fase di titolazione) o di 4 settimane (fase di mantenimento).
Schema di somministrazione
La prima iniezione deve essere somministrata sotto la supervisione di un operatore sanitario qualificato. Le successive iniezioni di Enspryng possono essere somministrate a casa propria da un paziente adulto/da una persona di assistenza, se il medico curante lo ritiene appropriato e se l'adulto/la persona di assistenza è padrone della tecnica di iniezione.
Se il paziente sviluppa i sintomi di una grave reazione allergica, i pazienti /le persone di assistenza devono immediatamente consultare un medico e informarsi presso il medico curante sulla possibilità di proseguire o meno il trattamento con Enspryng.
Modo di somministrazione
Iniezione sottocutanea (s.c.)
Enspryng deve essere somministrato con iniezione sottocutanea.
I siti di iniezione sono la parete addominale e la coscia. I siti di iniezione devono essere alternati, e le iniezioni non dovranno mai essere effettuate su nei, cicatrici o in zone cutanee sensibili alla pressione, lese, arrossate, indurite o comunque non integre.
Istruzioni dettagliate sulla somministrazione di Enspryng sono consultabili nelle «indicazioni per la manipolazione».
Controindicazioni
Enspryng è controindicato in pazienti con ipersensibilità nota a satralizumab o a una delle sostanze ausiliarie.
Avvertenze e misure precauzionali
Infezioni
Nei pazienti che presentano un'infezione in atto, la somministrazione di Enspryng deve essere rinviata a un momento successivo, fino a quando l'infezione non sia regredita (cfr. rubrica «Posologia/impiego - Somministrazione ritardata o saltata della dose»). I pazienti con infezioni recidivanti o affetti da malattie di base che favoriscono la comparsa di infezioni (ad es. diverticolite, diabete e pneumopatie interstiziali) devono essere trattati con prudenza.
Immunosoppressione
Durante il trattamento con Enspryng, la risposta immunitaria umorale può risultare compromessa.
Tubercolosi
Si raccomanda, come anche per le altre terapie immunomodulanti, prima della terapia con Enspryng, di esaminare i pazienti per escludere una eventuale infezione tubercolare latente. I pazienti con tubercolosi latente, prima di iniziare la terapia con Enspryng, devono essere sottoposti a una terapia antimicobatterica standard.
Vaccinazioni
Insieme a Enspryng non devono essere somministrati vaccini vivi o vaccini vivi attenuati, poiché in questi casi non è dimostrata la sicurezza clinica. L'intervallo di tempo tra la somministrazione di vaccini vivi e l'inizio della terapia con Enspryng deve corrispondere alle attuali linee guida sulle vaccinazioni riferibili ai principi attivi immunomodulanti/immunosoppressori.
Non sono disponibili dati sugli effetti di una vaccinazione in pazienti che ricevono Enspryng. In tutti i pazienti, prima di iniziare la terapia con Enspryng, si raccomanda di aggiornare tutte le vaccinazioni in concordanza con le attuali linee guida sulle vaccinazioni.
Enzimi epatici
Durante il trattamento con Enspryng sono stati osservati aumenti lievi e moderati delle transaminasi epatiche, in prevalenza inferiori a 5 volte il limite superiore della normalità (LSN), non limitanti il trattamento e reversibili durante la somministrazione di Enspryng.
Nei primi 3 mesi di trattamento, i valori di ALT e AST devono essere controllati ogni 4 settimane, in seguito per un anno ogni 3 mesi e successivamente in base all'indicazione clinica. Per le raccomandazioni sull'interruzione del trattamento, cfr. rubrica «Posologia/impiego - Aggiustamento della dose a causa di effetti indesiderati».
Riattivazione di epatite B
È stata riportata la riattivazione dell'epatite B in correlazione con le terapie biologiche. I pazienti con test positivo all'epatite sono stati esclusi dagli studi clinici con Enspryng.
Conta dei neutrofili
In seguito al trattamento con Enspryng si sono verificate riduzioni del numero di neutrofili. Una neutropenia di grado 3 e 4 si è manifestata nel 9,6% dei pazienti trattati con Enspryng e nel 5,4% dei pazienti trattati con placebo (cfr. rubrica «Effetti indesiderati»).
La conta dei neutrofili dovrà essere controllata 4-8 settimane dopo l'inizio del trattamento, e anche successivamente in base all'indicazione clinica. Per raccomandazioni relative all'interruzione della terapia cfr. rubrica «Posologia/impiego».
Abuso di medicamenti e dipendenza
Non sono stati condotti studi sull'abuso di medicamenti né sulla dipendenza. Dai dati disponibili non si evince che il trattamento con Enspryng possa determinare dipendenza.
In caso di passaggio a un'altra forma di somministrazione e/o a un altro medicamento con lo stesso principio attivo si raccomanda cautela. Il paziente deve essere opportunamente controllato.
Neoplasie maligne
I medicamenti immunomodulanti possono aumentare il rischio di tumori maligni.
Non è noto l'effetto del trattamento con Enspryng sullo sviluppo di patologie maligne.
Reazioni di ipersensibilità
Nei pazienti in trattamento con biologici possono manifestarsi reazioni di ipersensibilità. Prima di iniziare il trattamento, occorre informare i pazienti sui possibili sintomi di una reazione di ipersensibilità.
La prima iniezione deve essere somministrata sotto la supervisione di un operatore sanitario qualificato.
Al manifestarsi di reazioni di ipersensibilità i pazienti devono immediatamente informarne il loro medico ed eventualmente recarsi presso il suo studio e cercare assistenza d'emergenza.
Eventi cardiovascolari
Gli inibitori del recettore dell'IL 6 possono aumentare il rischio di malattie cardiovascolari. Pertanto, devono essere regolarmente controllati in particolare i pazienti con fattori di rischio quali ipertensione arteriosa, dislipidemia e diabete mellito (ECG, misurazione della pressione arteriosa).
Parametri lipidici
Nei pazienti trattati con Enspryng sono stati osservati aumenti dei trigliceridi e del colesterolo (parametri lipidici) (cfr. rubrica «Effetti indesiderati»). I pazienti con aumento dei parametri lipidici devono essere trattati in base alle attuali linee guida cliniche per il trattamento dell'iperlipidemia.
Attivazione del sistema del complemento
Durante il trattamento con inibitori del recettore dell'IL-6 si può verificare l'attivazione del sistema del complemento. Sulla base dei dati clinici finora disponibili, non è stato osservato un tale effetto durante il trattamento con Enspryng. A causa dei dati limitati, tuttavia, un tale rischio per Enspryng non può essere ancora valutato con sicurezza.
Malattie demielinizzanti del SNC
Durante il trattamento con inibitori del recettore dell'IL-6 è stata osservata la comparsa di altre malattie infiammatorie del SNC. Occorre verificare l'eventuale presenza di sintomi indicativi di una malattia demielinizzante di nuova comparsa. A causa dei dati limitati, tuttavia, un tale rischio per Enspryng non può essere ancora valutato con sicurezza.
Perforazione di diverticoli/perforazione intestinale
Durante il trattamento dell'artrite reumatoide (AR) con inibitori del recettore dell'IL-6 è stata osservata la comparsa di perforazione diverticolare o intestinale. Dagli studi cardine su Enspryng sono stati esclusi i pazienti con anamnesi nota di diverticolite. Non si può escludere durante il trattamento con Enspryng un aumentato rischio di perforazione diverticolare o intestinale, come succede per gli altri inibitori del recettore dell'IL-6. Nei pazienti con anamnesi di ulcere o diverticolite intestinale, Enspryng deve essere usato solo con cautela. Alla comparsa di dolori addominali acuti, i pazienti devono essere visitati immediatamente per poter riconoscere tempestivamente una eventuale perforazione gastrointestinale.
Interazioni
Con Enspryng non sono stati condotti studi formali di interazione farmacologica.
Interazioni farmacocinetiche
L'espressione degli enzimi epatici CYP450 viene soppressa dalle citochine, ad es. dalle citochine IL-6, che stimolano le infiammazioni croniche. Pertanto, l'espressione del CYP450 potrebbe cambiare quando viene indotta una inibizione delle citochine con Enspryng.
Per questo motivo, i pazienti che assumono medicamenti le cui dosi devono essere determinate individualmente e che sono metabolizzati da CYP450 3A4, 1A2 o 2C9 (ad es. atorvastatina, calcio-antagonisti, teofillina, warfarin, fenitoina, ciclosporina o benzodiazepine), devono essere monitorati all'inizio e al termine di una terapia con Enspryng per adattare, se necessario, le dosi di queste sostanze. In considerazione della sua lunga emivita di eliminazione, dopo il termine del trattamento, l'effetto di Enspryng sull'attività degli enzimi del CYP450 può persistere per varie settimane.
In analisi di farmacocinetica di popolazione non sono stati riscontrati effetti di AZA, corticosteroidi o MMF sulla clearance di Enspryng.
Gravidanza/Allattamento
Gravidanza
Non sono disponibili dati sull'uso di Enspryng in donne in gravidanza.
Gli studi condotti sulle scimmie non evidenziavano una tossicità diretta con effetti sulla gravidanza, né sullo sviluppo del feto né sullo sviluppo postnatale. Negli animali, satralizumab passa la barriera placentare e nella prole degli animali trattati con satralizumab, alcuni esiti potrebbero essere correlati con l'azione farmacologica sull'IL-6 (cfr. rubrica «Dati preclinici - Tossicità per la riproduzione»). La rilevanza clinica di questi risultati non è nota.
Enspryng non deve essere somministrato durante la gravidanza, a meno che il beneficio potenziale per la madre non superi il potenziale rischio per il feto. Poiché satralizumab passa la barriera placentare, nei bambini di madri trattate con Enspryng potrebbe sussistere un aumentato rischio di infezioni. Occorre prudenza nella somministrazione di vaccini vivi a questi bambini.
Nelle donne in età fertile deve essere preso in considerazione l'uso di un adeguato metodo contraccettivo, sia durante il trattamento sia fino a 5 mesi dopo l'ultima dose di Enspryng.
Allattamento
Non è noto se Enspryng sia secreto nel latte materno umano o se sia assorbito a livello sistemico dopo l'assunzione. Le donne trattate con Enspryng non devono allattare perché le IgG vengono secrete nel latte materno umano, e a livello preclinico la secrezione nel latte umano è stata dimostrata (cfr. rubrica «Dati preclinici»). Occorre valutare il beneficio dell'allattamento per il bambino in confronto al beneficio della terapia per la madre.
Fertilità
Non sono disponibili dati clinici sull'effetto di Enspryng sulla fertilità umana. Gli studi sperimentali sugli animali non hanno rivelato una compromissione della fertilità maschile o femminile (cfr. rubrica «Dati preclinici»).
Effetti sulla capacità di condurre veicoli e sull'impiego di macchine
Non sono stati effettuati studi riguardo agli effetti sulla capacità di guidare veicoli o utilizzare macchine. Dai dati disponibili, tuttavia, non è riconoscibile se il trattamento con Enspryng interferisce con la capacità di guidare veicoli o di utilizzare macchine.
Effetti indesiderati
Riassunto del profilo di sicurezza
La sicurezza di Enspryng in monoterapia o in associazione con una terapia immunosoppressiva è stata valutata in base ai dati provenienti da due studi clinici di fase III randomizzati, multicentrici, in doppio cieco, controllati con placebo (BN40898 e BN40900), nei quali 63 pazienti hanno ricevuto una monoterapia con Enspryng e 41 pazienti una terapia con Enspryng in associazione con una terapia immunosoppressiva (cfr. rubrica «Proprietà /effetti - Efficacia clinica»).
Gli effetti indesiderati (EI) più frequentemente riportati erano cefalea, artralgia, leucopenia e reazioni correlate all'iniezione.
I dati di sicurezza sono limitati in considerazione del numero di pazienti esposti a Enspryng e della durata dell'esposizione. Esiste la possibilità che eventuali effetti indesiderati, relativamente rari e potenzialmente gravi non siano stati evidenziati nel programma di studio. Non si possono escludere possibili effetti di classe in associazione con altri inibitori del recettore di IL-6, come ad es. perforazione intestinale, infezioni opportunistiche compresa la tubercolosi, riattivazione dell'epatite B, aumento del rischio cardiovascolare, attivazione del sistema del complemento, malattie demielinizzanti e tumori maligni (cfr. rubrica «Avvertenze e misure precauzionali»).
Tabella riassuntiva degli effetti indesiderati da farmaci osservati negli studi clinici
In tabella 1 sono riassunti gli EI riportati in correlazione con l'uso di Enspryng in monoterapia o in associazione con una terapia immunosoppressiva durante gli studi clinici. In entrambi gli studi clinici, i pazienti dei gruppi Enspryng sono stati trattati più a lungo rispetto ai pazienti del gruppo placebo (o del gruppo nel quale il placebo veniva associato con una terapia immunosoppressiva). Gli effetti indesiderati da farmaci osservato negli studi clinici (tabella 1) sono riportati in base alla classificazione per sistemi e organi secondo MedDRA. La categoria di frequenza relativa a ogni effetto indesiderato da farmaci è riportata sulla base della seguente convenzione: molto comune (≥1/10), comune (da ≥1/100 a <1/10), non comune (da ≥1/1'000 a <1/100), raro (da ≥1/10'000 a <1/1'000), molto raro (<1/10'000).
Tabella 1: riassunto degli effetti indesiderati in pazienti trattati nel contesto di studi clinici con Enspryng in monoterapia o in associazione con una terapia immunosoppressiva
Effetti indesiderati (MedDRA) | Quota pazienti (%) | Categoria di frequenza per Enspryng | |
Enspryng | Placebo1 | ||
Infezioni ed infestazioni | |||
Infezione delle vie urinarie | 18 (17,3%) | 15 (20,3%) | Molto comune |
Infezione delle vie respiratorie superiori | 20 (19,2%) | 12 (16,2%) | Molto comune |
Rinofaringite | 19 (18,3%) | 8 (10,8%) | Molto comune |
Influenza | 5 (4,8%) | 6 (8,1%) | Comune |
Patologie del sistema nervoso | |||
Cefalea | 20 (19,2%) | 8 (10,8%) | Molto comune |
Emicrania | 4 (3,8%) | 0 | Comune |
Traumatismo, avvelenamento e complicazioni da procedura | |||
Reazioni correlate all'iniezione | 13 (12,5%) | 7 (9,5%) | Molto comune |
Patologie del sistema muscoloscheletrico e del tessuto connettivo | |||
Artralgia | 14 (13,5%) | 1 (1,4%) | Molto comune |
Rigidità dell'apparato locomotore | 5 (4,8%) | 0 | Comune |
Patologie della cute e del tessuto sottocutaneo | |||
Eruzione cutanea | 9 (8,7%) | 3 (4,1%) | Comune |
Prurito | 6 (5,8%) | 1 (1,4%) | Comune |
Disturbi psichiatrici | |||
Insonnia | 6 (5,8%) | 1 (1,4%) | Comune |
Patologie generali e condizioni relative alla sede di somministrazione | |||
Edema periferico | 5 (4,8%) | 0 | Comune |
Patologie del sistema emolinfopoietico | |||
Ipofibrinogenemia | 3 (2,9%) | 0 | Comune |
Patologie respiratorie, toraciche e mediastiniche | |||
Rinite allergica | 4 (3,8%) | 0 | Comune |
Disturbi del metabolismo e della nutrizione | |||
Iperlipidemia | 14 (13,5%) | 9 (12,2%) | Molto comune |
Esami diagnostici | |||
Leucopenia | 14 (13,5%) | 4 (5,4) | Molto comune |
Iperbilirubinemia | 2 (1,9%) | 0 | Comune |
Aumento del peso corporeo | 5 (4,8%) | 2 (2,7%) | Comune |
Trombopenia | 4 (3,8%) | 2 (2,7%) | Comune |
Aumento delle transaminasi | 7 (6,7%) | 6 (8,1%) | Comune |
1 Placebo o placebo in associazione con una terapia immunosoppressiva
Descrizione di alcuni effetti collaterali selezionati
Reazioni correlate all'iniezione (RCI)
Le RCI riferite in pazienti trattati con Enspryng in monoterapia o in associazione con una terapia immunosoppressivo, sono risultate prevalentemente da lievi a moderate e si sono manifestate generalmente entro 24 ore dalle iniezioni. I sintomi sistemici riportati più frequentemente sono stati diarrea e cefalea. Le reazioni locali nel sito dell'iniezione più frequentemente riportate sono state sensazione di calore, eritema, prurito, eruzione cutanea e dolore. Nessuna delle reazioni correlate all'iniezione ha richiesto una interruzione o una sospensione della terapia.
Infezioni
Per tenere conto della maggiore durata di trattamento nel gruppo satralizumab rispetto al gruppo placebo, i dati sulle infezioni non sono riportati soltanto in percentuali, ma anche in eventi per 100 anni-paziente (eventi che si manifestano in 100 pazienti dopo 1 anno di trattamento).
Nello studio svolto con Enspryng in monoterapia, la frequenza delle infezioni insorte nei pazienti trattati con Enspryng risultava di 99,8 eventi/100 anni-paziente (IC 95%: 82,4; 119,8) e di 34/63 pazienti [54,0% (IC 95%: 40,94%; 66,61%)] rispetto a 162,6 eventi/100 anni-paziente (IC 95%: 125,8; 206,9) e 14/32 pazienti [43,8% (IC 95%: 26,36%; 62,34%)] nei pazienti del gruppo placebo. La frequenza delle infezioni gravi risultava di 5,2 eventi/100 anni-paziente (IC 95%: 1,9; 11,3) e di 6/63 pazienti [9,5% (IC 95%: 3,58%; 19,59%)] nei pazienti trattati con Enspryng rispetto a 9,9 eventi/100 anni-paziente (IC 95%: 2,7; 25,2) e 3/32 pazienti [9,4% (IC 95%: 1,98%; 25,02%)] nei pazienti del gruppo placebo.
Nei pazienti in trattamento con Enspryng in associazione con una terapia immunosoppressiva (TIS), la frequenza delle infezioni era di 132,5 eventi/100 anni-paziente (IC 95%: 108,2; 160,5) e di 28/41 pazienti [68,3% (IC 95%: 51,91%; 81,92%)] rispetto a 149,6 eventi/100 anni-paziente (IC 95%: 120,1; 184,1) e 26/42 pazienti [61,9% (IC 95%: 45,64%; 76,43%)] nei pazienti che ricevevano un placebo associato a TIS. La frequenza di infezioni gravi era di 2,6 eventi/100 anni-paziente (IC 95%: 0,3; 9,2) e di 2/41 pazienti [4,9% (IC 95%: 0,60%; 16,53%)] rispetto a 5,0 eventi/100 anni-paziente (IC 95%: 1,0; 14,7) e 3/42 pazienti [7,1% (IC 95%: 1,50%; 19,48%)] tra i pazienti che ricevevano placebo associato a TIS.
Aumento del peso corporeo
Nell'arco di tempo del trattamento in doppio cieco è stato osservato un aumento del peso corporeo uguale o superiore al 15% rispetto al valore iniziale nel 4,8% nei pazienti trattati con Enspryng (in monoterapia o in associazione con una terapia immunosoppressiva) e nel 2,7% dei pazienti che hanno ricevuto un placebo (o un placebo in associazione con una terapia immunosoppressiva).
Anomalie di laboratorio
Neutrofili
È stata rilevata una diminuzione del numero dei neutrofili durante la fase di trattamento in doppio cieco nel 31,7% dei pazienti trattati con Enspryng (in monoterapia o in associazione con una terapia immunosoppressiva) e nel 21,6% dei pazienti trattati con placebo (da solo o in associazione con una terapia immunosoppressiva). Nella maggior parte dei casi, la diminuzione del numero dei neutrofili è risultata transitoria o intermittente.
Tra i pazienti nel braccio Enspryng, il 9,6% presentava un valore dei neutrofili inferiore a 1 x 109/l, rispetto al 5,4% nel braccio placebo o nel braccio placebo con una terapia immunosoppressiva; questo dato non coincideva cronologicamente con gravi infezioni.
Trombociti
È stata rilevata una diminuzione del numero dei trombociti durante la fase di trattamento in doppio cieco nel 24,0% dei pazienti trattati con Enspryng (in monoterapia o in associazione con una terapia immunosoppressiva) e nel 9,5% dei pazienti trattati con placebo (da solo o in associazione con una terapia immunosoppressiva). La diminuzione del numero dei trombociti non coincideva con eventi emorragici.
La diminuzione del numero dei trombociti era transitoria e non risultava inferiore a 75 x 109/l. In nessun paziente il numero dei trombociti scendeva a ≤50 x 109/l.
Enzimi epatici
Sono stati rilevati aumenti dei valori di AST e ALT durante la fase di trattamento in doppio cieco rispettivamente nel 27,9% e nel 18,3% dei pazienti trattati con Enspryng (in monoterapia o in associazione con una terapia immunosoppressiva) e rispettivamente nel 12,2% e nel 13,5% dei pazienti che hanno ricevuto il placebo (da solo o associato a una terapia immunosoppressiva). Gli aumenti generalmente si collocavano a livelli inferiori al triplo del limite superiore della norma, erano transitori e reversibili senza interruzione del trattamento con Enspryng.
Aumenti dell'ALT o dell'AST superiori al triplo del valore superiore della norma si sono presentati rispettivamente nel 2,9% e nel 1,9% dei pazienti trattati con Enspryng in monoterapia o in associazione con una terapia immunosoppressiva, ma non sono stati accompagnati da un aumento della bilirubina totale. In un paziente trattato con Enspryng in associazione con una terapia immunosoppressiva sono stati misurati, 4 settimane dopo l'inizio della terapia, aumenti dell'ALT superiori a 5 volte il limite superiore della normalità, che si sono normalizzati dopo la sospensione di Enspryng.
Valori lipidici
Sono stati misurati aumenti del colesterolo totale a livelli superiori a 7,75 mmol/l durante la fase di trattamento in doppio cieco nel 10,6% dei pazienti trattati con Enspryng (in monoterapia o in associazione con una terapia immunosoppressiva) e nell'1,4% dei pazienti trattati con placebo (da solo o in associazione con una terapia immunosoppressiva). Nel 18,3% dei pazienti trattati con Enspryng si sono manifestati aumenti dei trigliceridi a livelli superiori ai 3,42 mmol/l, rispetto ai 6,8% dei pazienti del gruppo placebo. Gli aumenti dei valori lipidici non hanno richiesto l'interruzione della somministrazione.
La notifica di effetti collaterali sospetti dopo l'omologazione del medicamento è molto importante. Consente una sorveglianza continua del rapporto rischio-beneficio del medicamento. Chi esercita una professione sanitaria è invitato a segnalare qualsiasi nuovo o grave effetto collaterale sospetto attraverso il portale online ElViS (Electronic Vigilance System). Maggiori informazioni sul sito www.swissmedic.ch.
Posologia eccessiva
Non sono stati riportati casi di posologia eccessiva nei pazienti affetti da NMO o NMOSD. Non sono disponibili esperienze con posologia eccessiva nei pazienti affetti da NMO o NMOSD.
Segni e sintomi
In uno studio di fase I, volontari adulti sani hanno ricevuto con somministrazione sottocutanea una singola dose fino a 240 mg di Enspryng, senza che siano comparsi eventi indesiderati gravi.
Trattamento
In caso di sovradosaggio il paziente deve essere strettamente monitorato e deve ricevere un trattamento adeguato in base ai sintomi. Se necessario, occorre prendere misure di supporto.
Proprietà/Effetti
Codice ATC:
L04AC19
Meccanismo d'azione
Satralizumab è un anticorpo monoclonale IgG2 umanizzato (MAB), che si lega ai recettori umani di membrana e solubili dell'IL-6 (IL-6R), impendendo la trasmissione del segnale mediata da IL-6 attraverso questi recettori.
L'IL-6 è una citochina pleiotropica che viene prodotta da molti tipi di cellula e che partecipa a vari processi infiammatori, per esempio all'attivazione delle cellule B, alla differenziazione delle cellule B a plasmablasti e alla produzione di autoanticorpi, all'attivazione e differenziazione delle cellule Th17, all'inibizione delle cellule T regolatorie e alla modifica della permeabilità della barriera emato-encefalica. Durante le fasi di malattia attiva risultano aumentati i livelli di IL-6 nel liquor e nel siero dei pazienti affetti da NMO e NMOSD. Alcune delle funzioni dell'IL-6 vengono messe in correlazione con la patogenesi della NMO e delle NMOSD, tra cui la formazione di autoanticorpi patologici diretti contro l'aquaporina-4 (AQP4), una proteina del canale idrico, espressa principalmente dagli astrociti del SNC.
Farmacodinamica
Durante gli studi clinici svolti con Enspryng nella NMO e nelle NMOSD sono stati riscontrati riduzioni dei valori della proteina C reattiva (CRP), del fibrinogeno e del complemento (C3, C4 e CH50).
Efficacia clinica
L'efficacia e la sicurezza di Enspryng sono state valutate in due studi clinici cardine di fase III (BN40898 e BN40900) in pazienti affetti da NMO diagnosticata AQP4-IgG-sieropositiva o sieronegativa (criteri di Wingerchuk 2006) o da NMOSD diagnosticate AQP4-IgG-sieropositive (criteri di Wingerchuk 2007). In retrospettiva, questi pazienti soddisfacevano anche i criteri più nuovi proposti dal comitato internazionale per la diagnosi di NMO (rif: Wingerchuk et al. 2015). Gli effetti di Enspryng sono stati studiati in pazienti adulti (negli studi BN40898 e BN40900) e in pazienti adolescenti (con età dai 12 anni fino ai 18 anni, complessivamente N=7 (N=4 Enspryng, N=3 Placebo) nello studio BN40898). In entrambi gli studi, l'inclusione di pazienti adulti affetti da NMO, sieronegativi per AQP4-IgG, era limitata circa al 30% in modo tale che il collettivo di studio rispecchiasse il reale collettivo dei pazienti affetti da NMO.
In entrambi gli studi, l'indicatore primario di efficacia era il numero di recidive definite dal protocollo (PDR, protocol-defined relapse), basato su un peggioramento precedentemente stabilito dei risultati dell'EDSS (Expanded Disability Status Scale) e del FSS (Functional System Scores), confermato da un comitato indipendente per gli endpoint clinici CEC (Clinical Endpoint Committee). L'endpoint primario per l'analisi era la durata di tempo alla prima recidiva confermata dal CEC con valutazione EDSS/FSS entro 7 giorni dalla segnalazione dei sintomi da parte del paziente (recidiva aggiudicata).
Studio BN40898 (rispettivamente SA-307JG e SAkuraSky)
Lo studio BN40898 è stato uno studio clinico randomizzato, multicentrico, in doppio cieco, controllato con placebo, per la valutazione dell'efficacia di Enspryng in associazione con una terapia immunosoppressiva stabile (CO fino a 15 mg/giorno [equivalente a prednisolone], AZA fino a 3 mg/kg/die o MMF fino a 3'000 mg/die; gli adolescenti hanno ricevuto una associazione di AZA e CO o MMF e CO). Hanno partecipato allo studio 83 pazienti AQP4-IgG-sieropositivi e sieronegativi (tra cui 7 adolescenti). Durante le prime 4 settimane, i pazienti hanno ricevuto ogni 2 settimane le prime 3 dosi singole di 120 mg di Enspryng o un placebo tramite iniezione sottocutanea nella parete addominale o nella coscia e in seguito, una volta ogni 4 settimane.
Il disegno dello studio e le caratteristiche al basale del collettivo di studio sono rappresentati in tabella 2.
Lo studio è stato guidato dagli eventi e la fase di studio in doppio cieco per la valutazione dell'efficacia ha avuto termine dopo la comparsa di 26 recidive aggiudicate. Nella fase di estensione in aperto (open-label extension – OLE), durante la quale tutti i pazienti sono stati trattati con Enspryng in aperto, potevano essere accolti i pazienti che durante la fase in doppio cieco (DB) hanno presentato una recidiva (PDR) confermata dal CEC, che a causa della recidiva hanno ricevuto una terapia di salvataggio o che hanno concluso la fase in doppio cieco.
Tabella 2: Disegno dello studio e caratteristiche al basale dello studio BN40898
Nome dello studio | Studio BN40898 (N=83) | |
Disegno dello studio | ||
Popolazione dello studio | Pazienti adolescenti e adulti con NMO o NMOSD in trattamento stabile con una terapia immunosoppressiva. Età tra 12 e 74 anni, ≥2 recidive negli ultimi 2 anni prima dello screening (con almeno 1 recidiva nei 12 mesi prima dello screening), EDSS 0 - 6,5 | |
Durata dello studio fino alla valutazione dell'efficacia | Guidata dagli eventi (26 recidive definite nel protocollo di studio e confermate dal CEC) Durata mediana del follow-up: Enspryng 100 settimane, placebo 74 settimane | |
Gruppi di trattamento, randomizzazione 1:1 | Gruppo A: 120 mg di Enspryng s.c. Gruppo B: Placebo | |
Caratteristiche al basale | Enspryng + terapia immunosoppressiva | Placebo + terapia immunosoppressiva |
Diagnosi, n (%): |
|
|
NMO NMOSD | 33 (80,5) 8 (19,5) | 28 (66,7) 14 (33,3) |
Stato sieropositivo per AQP4-IgG, n (%) | 27 (65,9) | 28 (66,7) |
Età media in anni (deviazione standard) | 40,8 (16,1) (13 – 73) | 43,4 (12,0) (14 – 65) |
Adolescenti (da ≥12 a <18 anni), n (%) | 4 (9,8) | 3 (7,1) |
Distribuzione per sesso, |
|
|
n (%) maschili/ n (%) femminili | 4 (9,8) / 37 (90,2) | 2 (4,8) / 40 (95,2) |
Terapia immunosoppressiva, n (%): |
|
|
Corticosteroidi orali (CO) Azatioprina (AZA) Micofenolato mofetile (MMF) AZA + CO* MMF + CO* | 17 (41,5) 16 (39,0) 4 (9,8) 3 (7,3) 1 (2,4) | 20 (47,6) 13 (31,0) 8 (19,0) 0 1 (2,4) |
* Associazione approvata per pazienti adolescenti
Studio BN40900 (rispettivamente SA-309JG e SAkuraStar)
Lo studio BN40900 è stato uno studio clinico randomizzato multicentrico, in doppio cieco, controllato con placebo per valutare l'efficacia di una monoterapia con Enspryng al confronto con un placebo. Hanno partecipato allo studio 95 pazienti adulti AQP4-IgG-sieropositivi e sieronegativi. Durante le prime 4 settimane, i pazienti hanno ricevuto ogni 2 settimane le prime 3 dosi singole di 120 mg di Enspryng o un placebo tramite iniezione sottocutanea nella parete addominale o nella coscia e in seguito, una volta ogni 4 settimane.
Il disegno dello studio e le caratteristiche della popolazione di studio al basale sono rappresentati in tabella 3.
La fase di studio in doppio cieco per la valutazione dell'efficacia è terminata 1,5 anni dopo la data di randomizzazione dell'ultimo paziente accolto. È stato possibile accogliere nella fase OLE, durante la quale tutti i pazienti ricevevano in aperto il trattamento con Enspryng, i pazienti nei quali durante la fase in doppio cieco è comparsa una recidiva confermata dal CEC o che hanno concluso la fase in doppio cieco.
Tabella 3: Disegno dello studio e caratteristiche al basale dello studio BN40900
Nome dello studio | Studio BN40900 (N = 95) | |
Disegno dello studio | ||
Popolazione dello studio | Pazienti adulti con NMO o NMOSD Età 18-74 anni, ≥1 recidiva o primo evento negli ultimi 12 mesi prima dello screening, EDSS da 0 a 6,5. I pazienti avevano ricevuto un pretrattamento per prevenire una recidiva di NMOSD o non erano stati trattati. | |
Durata dello studio fino alla valutazione dell'efficacia | Guidata dagli eventi (44 recidive, definite dal protocollo di studio, confermate dal CEC o 1,5 anni dopo la data di randomizzazione dell'ultimo paziente ammesso, a seconda di ciò che si verifica per primo) Durata mediana del follow-up: Enspryng 95,4 settimane, Placebo 60,5 settimane | |
Gruppi di trattamento, randomizzazione 2:1 | Monoterapia: Gruppo A: 120 mg di Enspryng s.c. Gruppo B: placebo | |
Caratteristiche al basale | Enspryng (n = 63) | Placebo (n = 32) |
Diagnosi, n (%): |
|
|
NMO NMOSD | 47 (74,6) 16 (25,4) | 24 (75,0) 8 (25,0) |
Stato sieropositivo per AQP4-IgG, n (%) | 41 (65,1) | 23 (71,9) |
Età media in anni (deviazione standard) | 45,3 (12,0) (21 – 70) | 40,5 (10,5) (20 – 56) |
Distribuzione per sesso, |
|
|
n (%) maschili/ n (%) femminili | 17 (27,0) / 46 (73,0) | 1 (3,1) / 31 (96,9) |
Efficacia primaria - fase in doppio cieco
Nella popolazione ITT, il trattamento con Enspryng ha determinato una diminuzione del rischio di comparsa di una recidiva aggiudicata, statisticamente significativa, del 62% (Hazard Ratio [HR] [IC 95%]: 0,38 [0,16-0,88]; p [logrank test] = 0,0184) in caso di somministrazione in associazione con una terapia immunosoppressiva stabile (studio BN40898), e in caso di somministrazione in monoterapia è risultata una diminuzione del rischio di comparsa di una recidiva aggiudicata del 55% (HR [IC 95%]: 0,45 [0,23-0,89]; p [logrank test] = 0,0184) (studio BN40900), rispettivamente al confronto con il placebo.
Il più intenso effetto sottogruppo è stato osservato nei pazienti sieropositivi alle AQP4-IgG. Nei pazienti sieropositivi alle AQP4-IgG il rischio relativo di comparsa di una recidiva aggiudicata nello studio BN40898 è stato ridotto del 79% (HR [IC 95%]: 0,21 [0,06-0,75]) e nello studio BN40900 del 74% (HR [IC 95%: 0,26 [0,11-0,63]). Dopo 48 settimane, rispettivamente il 91,5% e l'82,9% dei pazienti sieropositivi alle AQP4-IgG, trattati con Enspryng in associazione con una terapia immunosoppressiva o in monoterapia sono rimasti senza recidive aggiudicate. Dopo 96 settimane, rispettivamente il 91,5% e il 76,5% dei pazienti sieropositivi alle AQP4-IgG trattati con Enspryng in associazione con una terapia immunosoppressiva o in monoterapia, sono rimasti senza recidive aggiudicate (cfr. tabella 4 e fig. 1 e 2). Combinando dati degli studi BN40898 e BN40900, il trattamento con Enspryng con o senza una terapia immunosoppressiva determinava nei pazienti sieropositivi alle AQP4-IgG una diminuzione del rischio complessivamente del 75% (HR [IC 95%]: 0,25 [0,12-0,50]). Le differenze riferibili alla durata del tempo alla prima recidiva aggiudicata nel gruppo dei pazienti sieronegativi alle AQP4-IgG tra quelli che hanno ricevuto Enspryng con o senza una terapia immunosoppressiva, e quelli che hanno ricevuto un placebo con o senza una terapia immunosoppressiva non risultavano statisticamente significative (BN40898 e BN40900 combinato HR [IC 95%]: 0,97 [0,41-2,33]).
Tabella 4: I più importanti endpoint di efficacia negli studi BN40898 e 40900 su pazienti sieropositivi alle AQP4-IgG
BN40898 | BN40900 | |||
Enspryng | Placebo + terapia | Enspryng | Placebo | |
Endpoint primario nei pazienti sieropositivi alle AQP4-IgG | ||||
Numero dei pazienti sieropositivi alle AQP4-IgG (n) | 27 | 28 | 41 | 23 |
Riduzione del rischio (studi singoli) | 79% (HR: 0,21; IC 95%: 0,06; 0,75; p = 0,0086) | 74% (HR: 0,26; IC 95%: 0,11; 0,63; p = 0,0014) | ||
Riduzione del rischio (analisi aggregata) | 75% (HR: 0,25; IC 95%: 0,12; 0,50; p: <0,0001) | |||
Quota dei pazienti senza recidiva aggiudicata dopo 48 settimane | 91,5% (IC 95%: 69,64; 97,83) | 59,9% (IC 95%: 36,25; 77,25) | 82,9% (IC 95%: 67,49; 91,47) | 55,4% (IC 95%: 32,96; 73,08) |
Quota dei pazienti senza recidiva aggiudicata dopo 96 settimane | 91,5% (IC 95%: 69,64; 97,83) | 53,3% (IC 95%: 29,34; 72,38) | 76,5% (IC 95%: 59,22; 87,21) | 41,1% (IC 95%: 20,76; 60,41) |
Fig. 1: Studio BN40898: tempo alla prima recidiva aggiudicata durante la fase in doppio cieco in pazienti sieropositivi alle AQP4-IgG

Fig. 2: Studio BN40900: tempo alla prima recidiva aggiudicata durante la fase in doppio cieco in pazienti sieropositivi alle AQP4-IgG

Il trattamento dei pazienti sieropositivi alle AQP4-IgG con Enspryng ha ridotto il tasso annualizzato delle recidive aggiudicate (Adjudicated Relapse Rate – ARR) rispetto al placebo nello studio BN40898 dell'88% e nello studio BN40900 del 90% (tabella 5).
Tabella 5: Tasso annualizzato delle recidive aggiudicate durante la fase in doppio cieco (modello di regressione binomiale negativa)
BN40898 | BN40900 | Dati aggregati | |||||
Placebo | Enspryng | Placebo | Enspryng | Placebo | Enspryng | ||
Pazienti sieropositivi alle AQP4-IgG | N = 28 | N = 27 | N = 23 | N = 41 | N = 51 | N = 68 | |
Pazienti con recidiva | 12 | 3 | 13 | 9 | 25 | 12 | |
Tasso annualizzato di recidiva rettificato | 0,520 | 0,063 | 2,853 | 0,275 | 1,339 | 0,136 | |
Riduzione ARR relativa (rapporto tra tassi) | 88% (RR: 0,122; IC 95%: 0,027; 0,546; p=0,0039) | 90% (RR: 0,096; IC 95%: 0,020; 0,473; p= 0,0086) | 90% (RR: 0,102; IC 95%: 0,034; 0,301; p=0,0002) | ||||
I pazienti sieropositivi alle AQP4-IgG trattati con Enspryng necessitano più raramente di una terapia di salvataggio (ad es. corticosteroidi, immunoglobuline e.v. e/o aferesi [compresa la plasmaferesi e lo scambio plasmatico]) rispetto ai pazienti trattati con placebo, e precisamente del 61% nello studio BN40898 e del 74% nello studio BN40900.
Tabella 6: Ricorso a una terapia di salvataggio nei pazienti con recidiva durante la fase in doppio cieco
BN40898 | BN40900 | Dati aggregati | ||||
Placebo | Enspryng | Placebo | Enspryng | Placebo | Enspryng | |
Pazienti sieropositivi alle AQP4-IgG | N = 28 | N = 27 | N = 23 | N = 41 | N = 51 | N = 68 |
Pazienti con terapia di salvataggio | 18 (64,29%) | 11 (40,74%) | 14 (60,87%) | 13 (31,71%) | 32 (62,75%) | 24 (35,29%) |
Riduzione del rischio (Odds Ratio) | 61% (OR: 0,3930; IC 95%: 0,1343; 1,1502 p=0,0883) | 74% (OR:0,2617; IC 95%: 0,0862; 0,7943; p=0,0180) | 66% (OR: 0,3430; IC 95%: 0,1614; 0,7289; p=0,0054) | |||
Nello studio BN40898 il trattamento dei pazienti sieropositivi alle AQP4-IgG con Enspryng, al confronto con il placebo ha ridotto il rischio di una grave recidiva (definita come aumento dell'EDSS di ≥2 punti rispetto alla precedente valutazione EDSS) dell'85% e nello studio BN40900 del 79% (tabella 7).
Tabella 7: Tempo alla prima recidiva aggiudicata durante la fase in doppio cieco
BN40898 | BN40900 | Dati aggregati | ||||
Placebo | Enspryng | Placebo | Enspryng | Placebo | Enspryng | |
Pazienti sieropositivi alle AQP4-IgG | N=27 | N=27 | N=23 | N=41 | N=50 | N=68 |
Pazienti con eventi | 6 (22,2%) | 1 (3,7%) | 5 (21,7%) | 3 (7,3%) | 11 (22,0%) | 4 (5,9%) |
Riduzione del rischio | 85% (HR: 0,15; IC 95%: 0,02; 1,25; p=0,0441) | 79% (HR: 0,21; IC 95%: 0,05; 0,91; p=0,0231) | 82% (HR: 0,18; IC 95%: 0,06; 0,58; p=0,0015) | |||
Il trattamento dei pazienti sieropositivi alle AQP4-IgG con Enspryng ha ridotto il rischio di peggioramento sulla scala EDSS rispetto al valore al basale nello studio BN40898 del 65% e nello studio BN40900 del 66% rispetto al trattamento con placebo (tabella 8).
Tabelle 8: Tempo al peggioramento dei punteggi sulla scala estesa per la valutazione dello status di disabilità (Expanded Disability Status Scale, EDSS)* nella fase in doppio cieco (pazienti sieropositivi alle AQP4-IgG)
BN40898 | BN40900 | Combinato | ||||||
Placebo | Enspryng | Placebo | Enspryng | Placebo | Enspryng | |||
Pazienti sieropositivi alle AQP4-IgG | N = 27 | N = 28 | N = 23 | N = 41 | N = 50 | N = 69 | ||
Pazienti con eventi | 12 (44,4%) | 5 (17,9%) | 11 (47,8%) | 11 (26,8%) | 23 (46,0%) | 16 (23,2%) | ||
Riduzione del rischio | 65% (HR: 0,35;IC 95%: 0,12; 1,00; p = 0,0407) | 66% (HR: 0,34;IC 95%: 0,14; 0,82; p = 0,0124) | 69% (HR: 0,31;IC 95%: 0,16; 0,59; p = 0,0002) | |||||
* Il peggioramento sulla scala EDSS rispetto al basale è stato definito come: a) peggioramento del punteggio EDSS di ≥2 punti nei pazienti con score iniziale di 0, b) peggioramento dello score EDSS di ≥1 punti nei pazienti con uno score iniziale da 1 a 5, c) peggioramento dello score EDSS di ≥0,5 punti nei pazienti con uno score iniziale di ≥5,5.
Fase di estensione in aperto
Dalle analisi dei dati a lungo termine, tra cui di quelli emersi dalla fase di estensione in aperto in corso (sulla base delle recidive trattate con terapia di salvataggio), è risultato che rispettivamente il 58% e il 73% dei pazienti sieropositivi alle AQP4-IgGr trattati con Enspryng restavano liberi da recidive dopo 120 settimane di trattamento, quando Enspryng è stato somministrato in associazione o in monoterapia.
Complessivamente sono disponibili solo dati limitati sull'efficacia e sulla sicurezza del trattamento con Enspryng per più di 2 anni.
Fig. 3: Studio BN40898: tempo alla prima recidiva (recidiva clinica trattata) durante la fase in doppio cieco e in aperto nei pazienti sieropositivi alle AQP4-IgG

Fig. 4: Studio BN40900: tempo alla prima recidiva (recidiva clinica trattata) durante la fase in doppio cieco e in aperto nei pazienti sieropositivi alle AQP4-IgG

Caratteristiche al basale ed efficacia nei pazienti adolescenti (studio BN40898)
L'età media dei 7 adolescenti accolti nello studio BN40898 durante la fase in doppio cieco, è stata di 15,4 anni, il peso corporeo mediano è stato di 79,6 kg. La maggior parte di questi pazienti adolescenti era di sesso femminile (n = 6). Quattro pazienti erano di etnia bianca, 2 pazienti di etnia nera/afroamericana, e 1 paziente di etnia asiatica. Tre dei sette (42,9%) pazienti adolescenti sono risultati sieropositivi alle AQP4-IgG (2 nel gruppo placebo e 1 nel gruppo Enspryng) allo screening. Nella fase in doppio cieco è comparsa una recidiva aggiudicata in 1 sui 3 pazienti adolescenti del gruppo placebo e in 1 sui 4 adolescenti nel gruppo Enspryng. A causa del limitato numero di casi non è stata calcolata, in questo sottogruppo di pazienti, la Hazard Ratio per l'endpoint primario, la durata alla prima recidiva aggiudicata.
Ulteriori informazioni
Immunogenicità
Nello studio di fase III BN40898 (associazione con una terapia immunosoppressiva) e nello studio di fase III BN40900 (monoterapia) sono stati riscontrati anticorpi anti-farmaco (anti-drug antibodies, ADA) rispettivamente nel 41% e nel 71% dei pazienti che nella fase in doppio cieco hanno ricevuto Enspryng. Non è noto in quale misura questi ADA siano in grado di neutralizzare il legame con Enspryng.
L'esposizione è stata inferiore nei pazienti ADA-positivi, tuttavia non vi sono stati effetti degli ADA sulla sicurezza e non vi sono stati effetti sull'efficacia o marker farmacodinamici indicativi di un legame con le molecole-bersaglio.
Negli studi di fase III, il trattamento con satralizumab ha determinato una diminuzione simile del rischio di comparsa di una recidiva aggiudicata nei pazienti, nonostante le diverse frequenze della presenza di ADA nei singoli casi. I pazienti con un peso corporeo maggiore e con esposizione minore sviluppavano con maggiore probabilità anticorpi antifarmaco (indipendentemente da un trattamento di base con una terapia immunosoppressiva), tuttavia, l'effetto del trattamento in tutti i gruppi di peso corporeo è risultato equiparabile, sia per la somministrazione in associazione con una terapia immunosoppressiva, sia in monoterapia.
La dose raccomandata è adatta a tutti i pazienti, e nei pazienti che sviluppano ADA non occorre né una interruzione della terapia né una modifica del dosaggio.
Farmacocinetica
La farmacocinetica di Enspryng è stata descritta in volontari sani sia giapponesi sia di etnia caucasica e in pazienti affetti da NMO e da NMOSD. La caratterizzazione della farmacocinetica in pazienti con NMO e NMOSD con l'uso della dose raccomandata è stata svolta in base ad analisi di farmacocinetica di popolazione.
Lo svolgimento della concentrazione in relazione al tempo di Enspryng nei pazienti con NMO o NMOSD è stato descritto con un modello di farmacocinetica di popolazione a due compartimenti con eliminazione parallela lineare e diretta all'obiettivo (cinetica di Michaelis-Menten) e assorbimento sottocutaneo (SC) di primo ordine. I parametri della clearance e del volume di Enspryng sono stati rappresentati in modo allometrico in scala in base al peso corporeo (con una funzione power con coefficiente fisso di power rispettivamente di 0,75 o 1 per i parametri di clearance e di volume). Il peso corporeo è risultato essere una covariata significativa, infatti la clearance e il Vc (volume di distribuzione centrale) aumentavano rispettivamente del 71,3% e del 105% nei pazienti con un peso corporeo di 123 kg (97,5-esimo percentile della distribuzione del peso) al confronto con un paziente con un peso corporeo di 60 kg.
La farmacocinetica allo stato stazionario è stata raggiunta dopo la fase di saturazione (8 settimane) con i seguenti valori per Cmin, Cmax e AUC (media (± DS*)): Cmin: 28,5 (10,2) µg/ml, Cmax: 42,7 (11,9) µg/ml e AUC: 1020 (313) µg.ml/giorno. (*deviazione standard) La farmacocinetica non è stata alterata dalla immunoterapia di base (cfr. rubrica «Interazioni”).
Assorbimento
La costante di velocità dell'assorbimento di Enspryng è stata di 0,251 1/giorno (IC 95%: 0,216-0,285), corrispondente all'emivita di assorbimento di circa 3 giorni, usando la dose raccomandata (cfr. rubrica «Posologia/impiego»). La biodisponibilità è risultata elevata (85,4%, IC 95%: 0,795-0,953).
Distribuzione
Enspryng presenta una distribuzione bifasica. Il volume di distribuzione centrale è stato di 3,46 l (IC 95%: 3,21-3,97), il volume di distribuzione periferico di 2,07 l (IC 95%: 1,78-2,59). La clearance intercompartimentale è stata di 0,336 l/giorno (IC 95%: 0,261-0,443).
Metabolismo
Il metabolismo di Enspryng non è stato studiato direttamente. Si presume che Enspryng, subisca una eliminazione catabolica.
Eliminazione
La clearance di Enspryng dipende dalla concentrazione. La clearance lineare è stimata di 0,0601 l/giorno (IC 95%: 0,0524-0,0695) e rappresenta circa la metà della clearance complessiva nello steady state usando la dose raccomandata nei pazienti con NMO e NMOSD. Sulla base di dati combinati emersi dagli studi di fase 3, il t1/2 terminale correlato è di circa 30 giorni (range 22-37 giorni).
Cinetica di gruppi di pazienti speciali
Nelle analisi di cinetica di popolazione su pazienti adulti affetti da NMO o NMOSD non sono stati riscontrati effetti dell'età, del sesso o dell'appartenenza etnica sulla farmacocinetica di satralizumab. Sebbene il peso corporeo abbia avuto un effetto sulla farmacocinetica di satralizumab, non sussistono raccomandazioni sull'adeguamento della dose per tenere conto di queste caratteristiche demografiche.
Disturbi della funzionalità epatica
Non è stato svolto nessuno studio formale sugli effetti della disfunzione epatica sulla farmacocinetica di satralizumab.
Disturbi della funzionalità renale
Non è stato svolto nessuno studio formale sugli effetti della disfunzione renale sulla farmacocinetica di satralizumab. Tuttavia, agli studi clinici BN40898 e BN40900 hanno partecipato 22 pazienti con lieve disfunzione renale (Clearance della creatinina rispettivamente di <80 ml/min e ≥50 ml/min). Come era stato atteso in base ai noti meccanismi di clearance di satralizumab, la farmacocinetica in questi pazienti non è risultata compromessa, e pertanto non occorre un adeguamento della dose.
Pazienti anziani
Non sono stati svolti studi specifici per valutare la farmacocinetica di satralizumab nei pazienti di età superiore ai 65 anni. Tuttavia, agli studi clinici BN40898 e BN40900 hanno partecipato pazienti affetti da NMO o NMOSD di età tra i 65 e i 74 anni.
Le analisi di farmacocinetica di popolazione basate sui dati di questi pazienti hanno dimostrato che l'età non svolge alcun effetto sulla farmacocinetica di satralizumab.
Bambini e adolescenti
I dati rilevati da 8 pazienti adolescenti [13-17 anni], nei quali è stato impiegato lo schema di somministrazione per adulti, dimostrano che i parametri di farmacocinetica di popolazione di satralizumab non divergono significativamente da quelli degli adulti.
Pertanto non è necessario un adeguamento della dose.
Dati preclinici
Farmacologia di sicurezza/tossicità cronica (o tossicità per somministrazione ripetuta)
Studi preclinici su scimmie, una specie responder con reattività crociata verso satralizumab, basati sulla farmacologia di sicurezza e sugli endpoint di tossicità non hanno evidenziato un particolare rischio per l'uomo. Non sono stati osservati effetti avversi con somministrazione sottocutanea, una volta alla settimana, di una dose fino a 50 mg di satralizumab/kg a scimmie Cynomolgus durante studi di tossicità di 4 e 26 settimane. L'unica modificazione rilevante durante questi studi è stato un aumento del livello di IL-6 nel sangue, che è stato valutato come risultato dell'effetto farmacologico di satralizumab (blocco di IL-6). Nella maggior parte degli animali trattati, satralizumab ha determinato una risposta immune con anticorpi anti-farmaco, che tuttavia non ha influito sulla risposta farmacologica e non ha determinato effetti indesiderati.
Genotossicità
Non sono stati svolti studi per il rilevamento del potenziale genotossico di satralizumab. Si presume che gli anticorpi non abbiano effetti sul DNA.
Cancerogenicità
Non sono stati svolti studi di cancerogenicità con satralizumab. In uno studio sulla tossicità cronica della durata di 6 mesi su scimmie Cynomolgus non sono state riscontrate lesioni proliferative.
Fertilità
La somministrazione a lungo termine di satralizumab nelle scimmie non ha avuto effetti sugli organi di riproduzione maschili o femminili.
Tossicità per la riproduzione
Il trattamento di scimmie Cynomolgus gravide con dosi fino a 50 mg di satralizumab/kg/sett. fino al parto non ha determinato effetti negativi sulle madri, sullo sviluppo dei feti, sull'esito della gravidanza, né sulla sopravvivenza e sullo sviluppo della figliata compresa la capacità di apprendimento. La reazione anticorpale a un antigene cellula-T-dipendente è stata più bassa nei discendenti di animali trattati con satralizumab rispetto a quella nei discendenti di animali di controllo. Probabilmente questo dato è in correlazione con l'effetto farmacologico sull'IL-6.
I discendenti di animali madri trattate con satralizumab presentavano una esposizione sistemica e fino a 6 mesi dopo la nascita presentavano livelli elevati di IL-6 nel plasma. Satralizumab è stato riscontrato nel latte (<0,9% del rispettivo livello plasmatico della madre).
Altre indicazioni
Stabilità
Il medicamento non dev'essere utilizzato oltre la data indicata con «EXP» sul contenitore.
Indicazioni particolari concernenti l'immagazzinamento
Conservare in frigorifero (2-8 °C).
Se necessario e se non aperto, Enspryng può essere prelevato dal frigorifero ed esservi riposto. Conservato a temperatura ambiente e senza refrigerazione (max. 30 °C) la durata di 8 giorni non deve essere superata.
Non scuotere. Non congelare.
Conservare il contenitore nella scatola originale per proteggere il contenuto dalla luce.
Conservare fuori dalla portata dei bambini.
Indicazioni per la manipolazione
Enspryng è destinato al monouso.
Prima dell'uso, conservare la siringa preriempita per 30 minuti fuori dalla scatola pieghevole a temperatura ambiente.
Non iniettare il medicamento se il liquido si presenta torbido o di colore anomalo o se contiene particelle.
Controllare la siringa preriempita e la protezione dell'ago per verificare che non siano presenti danneggiamenti. Non usare se sono presenti fessure o difetti.
Smaltimento della siringa preriempita e della protezione dell'ago.
Durante l'uso e lo smaltimento della siringa preriempita e della protezione dell'ago vanno assolutamente osservati i seguenti aspetti:
- la siringa preriempita non deve assolutamente essere riutilizzata.
- Subito dopo l'uso, porre la siringa usata in un contenitore di smaltimento per oggetti affilati e appuntiti.
- Smaltire la siringa preriempita e la protezione dell'ago secondo le normative locali in vigore o in base alle istruzioni degli operatori sanitari.
- Tenere la siringa preriempita, la protezione dell'ago e tutti i medicamenti fuori dalla portata dei bambini.
Smaltimento di medicamenti non usati/scaduti
La dispersione di medicamenti nell'ambiente deve essere ridotta al minimo. I medicamenti non devono essere smaltiti nelle acque di scarico. Lo smaltimento con i rifiuti domestici deve essere evitato. Usare «sistemi di raccolta» appropriati, se disponibili nella propria città.
Numero dell'omologazione
67617 (Swissmedic).
Titolare dell’omologazione
Roche Pharma (Svizzera) SA, Basilea.
Stato dell'informazione
Luglio 2020.
▼ Ce médicament fait l'objet d'une surveillance supplémentaire qui permettra l'identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave. Voir la rubrique «Effets indésirables» pour les modalités de déclaration des effets secondaires.
Composition
Principes actifs
Satralizumabum (produit par génie génétique à l'aide de cellules CHO [ovaire de hamster chinois]).
Excipients
L-histidinum; acidum L-asparticum; L-arginini hydrochloridum; poloxamera 188; aqua ad iniectabilia q.s. ad solutionem pro 1,0 ml.
Forme pharmaceutique et quantité de principe actif par unité
Solution injectable en seringue préremplie.
Solution stérile prête à l'emploi pour injection sous-cutanée (s. c.) dans une seringue préremplie munie d'un dispositif de protection de l'aiguille (Needle Safety Device, NSD) et contenant une dose unique.
La solution pour injection s. c. d'Enspryng est un liquide incolore à jaune pâle dans une seringue préremplie de 1 ml de solution. Chaque seringue préremplie contient 120 mg de satralizumab.
Indications/Possibilités d’emploi
Enspryng est utilisé en monothérapie ou en association avec un traitement immunosuppresseur (TIS) dans le traitement des maladies du spectre de la neuromyélite optique (NMOSD) chez les adultes et les adolescents présentant des anticorps IgG anti-aquaporine-4 (c.-à-d. séropositifs pour les IgG anti-AQP4).
Posologie/Mode d’emploi
Généralités
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Posologie recommandée
Enspryng doit être administré en injection sous-cutanée.
Instauration du traitement/Dose de charge
La dose de charge recommandée est de 120 mg en injection s. c. toutes les 2 semaines (première dose dans la semaine 0, deuxième dose dans la semaine 2 et troisième dose dans la semaine 4) lors des trois premières administrations.
Traitement d'entretien
La dose d'entretien recommandée est de 120 mg en injection s. c. toutes les 4 semaines (à partir de la semaine 8).
Durée du traitement
Enspryng est destiné au traitement à long terme.
Ajustement de la posologie du fait d'effets indésirables/d'interactions
Modifications des enzymes hépatiques
Le traitement par Enspryng doit être définitivement arrêté si l'élévation du taux d'alanine aminotransférase (ALAT) ou d'aspartate aminotransférase (ASAT) est supérieure à 5 fois la limite supérieure de la normale (LSN) et est associée à une élévation du taux de bilirubine.
Le traitement par Enspryng doit être interrompu si l'élévation du taux d'ALAT ou d'ASAT est supérieure à 5 fois la limite supérieure de la normale (LSN), mais n'est pas associée à une élévation du taux de bilirubine. Il peut être poursuivi (120 mg en injection s. c. toutes les 4 semaines), lorsque les taux d'ALAT et d'ASAT se sont normalisés et après évaluation du rapport bénéfice/risque du traitement pour le patient. Les paramètres hépatiques doivent être étroitement surveillés si l'on décide de poursuivre le traitement. Le médicament doit être définitivement arrêté si une nouvelle élévation des taux d'ASAT/ALAT et/ou de bilirubine est observée ultérieurement.
Neutropénie
Si le nombre de neutrophiles est inférieur à 1,0 x 109/l et qu'il est confirmé lors de plusieurs analyses, Enspryng doit être interrompu jusqu'à ce que le nombre de neutrophiles soit remonté à plus de 1,0 x 109/l.
Traitement associé
Enspryng peut être utilisé en monothérapie ou en association avec des corticostéroïdes oraux (CSO), l'azathioprine (AZA) ou le mycophénolate mofétil (MMF) (voir la rubrique «Propriétés/Effets, Efficacité clinique»). Il convient en outre de tenir compte des informations professionnelles de ces produits.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
La sécurité et l'efficacité d'Enspryng n'ont pas été évaluées chez les patients présentant des troubles de la fonction hépatique (voir la rubrique «Pharmacocinétique, Cinétique pour certains groupes de patients»).
Patients présentant des troubles de la fonction rénale
La sécurité et l'efficacité d'Enspryng n'ont pas fait l'objet d'études spécifiques chez les patients présentant des troubles de la fonction rénale. Toutefois, comme Enspryng est un anticorps monoclonal qui est soumis à des processus de dégradation dans l'organisme (et n'est pas éliminé par voie rénale), aucun ajustement posologique ne devrait être nécessaire chez les patients présentant des troubles de la fonction rénale (voir la rubrique «Pharmacocinétique, Cinétique pour certains groupes de patients»). Des patients présentant une insuffisance rénale légère ont été inclus dans les études cliniques et la pharmacocinétique du satralizumab n'a pas été modifiée chez ces patients (voir la rubrique «Pharmacocinétique, Cinétique pour certains groupes de patients»).
Patients âgés
La sécurité et l'efficacité d'Enspryng ont été évaluées chez des patients jusqu'à 74 ans. Aucun ajustement posologique n'est nécessaire chez les patients ≥65 ans (voir la rubrique «Pharmacocinétique, Cinétique pour certains groupes de patients»).
La sécurité et l'efficacité d'Enspryng n'ont pas été évaluées chez les patients de plus de 74 ans (voir la rubrique «Posologie/Mode d'emploi, Instructions posologiques particulières»).
Enfants et adolescents
La sécurité et l'efficacité d'Enspryng ont été évaluées chez un nombre limité (N=4) de patients adolescents ≥12 ans. Les résultats concernant la pharmacocinétique, l'efficacité et la sécurité étaient comparables à ceux obtenus chez les adultes (voir la rubrique «Propriétés/Effets, Efficacité clinique» et la rubrique «Pharmacocinétique, Cinétique pour certains groupes de patients»).
La sécurité et l'efficacité d'Enspryng n'ont pas été évaluées chez les patients pédiatriques de moins de 12 ans.
Prise retardée ou oubliée
Si une injection est omise, celle-ci doit être réalisée aussi rapidement que possible, sans attendre le moment prévu pour la dose suivante. Après l'administration de la dose reportée ou oubliée, il convient de respecter un intervalle de 2 semaines (phase de charge) ou de 4 semaines (phase d'entretien) entre les doses.
Schéma d'administration
La première injection doit être faite sous la surveillance d'un professionnel de la santé qualifié. Un patient adulte/un aidant peut faire les injections d'Enspryng suivantes à domicile si le médecin traitant le juge approprié et que le patient adulte/l'aidant maîtrise la technique d'injection.
Si le patient présente des symptômes d'une réaction allergique grave, le patient/l'aidant doit immédiatement faire appel à un médecin et demander au médecin traitant s'il est possible ou non de poursuivre le traitement par Enspryng.
Mode d'administration
Injection sous-cutanée (s. c.)
Enspryng doit être administré en injection sous-cutanée.
Les sites d'injection recommandés sont la paroi abdominale et la cuisse. Il faut alterner les sites d'injection et ne jamais faire les injections dans des grains de beauté, des cicatrices ou dans des zones où la peau est sensible à la pression, lésée, rouge, indurée ou non intacte.
Des instructions complètes relatives à l'administration d'Enspryng figurent dans la rubrique «Remarques concernant la manipulation».
Contre-indications
Enspryng est contre-indiqué chez les patients présentant une hypersensibilité connue au satralizumab ou à l'un des excipients.
Mises en garde et précautions
Infections
Chez les patients présentant une infection active, l'administration d'Enspryng doit être reportée à une date ultérieure jusqu'à ce que l'infection ait disparu (voir la rubrique «Posologie/Mode d'emploi, Prise retardée ou oubliée»). La prudence est recommandée chez les patients présentant des infections récurrentes et chez les patients souffrant de maladies sous-jacentes favorisant la survenue d'infections (p.ex. diverticulite, diabète et pneumopathie interstitielle).
Immunosuppression
La réponse immunitaire humorale peut être diminuée sous Enspryng.
Tuberculose
Comme cela est recommandé avec d'autres traitements immunomodulateurs, la présence d'une infection tuberculeuse latente doit être recherchée chez tous les patients avant d'instaurer un traitement par Enspryng. Les patients ayant une tuberculose latente doivent recevoir un traitement antimycobactérien standard avant l'instauration du traitement par Enspryng.
Vaccinations
Les vaccins vivants et les vaccins vivants atténués ne doivent pas être administrés en même temps qu'Enspryng, car la sécurité clinique n'est pas établie. L'intervalle entre l'administration de vaccins vivants et l'instauration du traitement par Enspryng doit être conforme aux recommandations vaccinales actuelles relatives aux principes actifs immunomodulateurs/immunosuppresseurs.
Il n'existe aucune donnée sur les effets d'une vaccination chez les patients recevant Enspryng. Avant d'instaurer le traitement par Enspryng, il est recommandé, chez tous les patients, de mettre à jour toutes les vaccinations conformément aux recommandations vaccinales actuelles.
Enzymes hépatiques
Des élévations légères et modérées des transaminases hépatiques ont été observées pendant le traitement par Enspryng. La majorité de ces élévations étaient inférieures à 5 fois la LSN, n'ont pas limité le traitement et ont été réversibles pendant l'administration d'Enspryng.
Les taux d'ALAT et d'ASAT doivent être contrôlés toutes les 4 semaines pendant les 3 premiers mois du traitement, puis tous les 3 mois pendant un an et par la suite, selon l'indication clinique. Concernant les recommandations relatives à l'arrêt du traitement, voir la rubrique «Posologie/mode d'emploi, Ajustement de la posologie du fait d'effets indésirables».
Réactivation d'une hépatite B
La réactivation d'une hépatite B a été rapportée en relation avec des traitements biologiques. Les patients ayant un test de dépistage de l'hépatite B positif ont été exclus des études cliniques menées avec Enspryng.
Nombre de neutrophiles
Une diminution du nombre de neutrophiles a été observée après le traitement par Enspryng. Une neutropénie de grade 3 et 4 est survenue chez 9,6% des patients sous Enspryng et chez 5,4% des patients sous placebo (voir la rubrique «Effets indésirables»).
Le nombre de neutrophiles doit être surveillé pendant 4 à 8 semaines après l'instauration du traitement, puis selon l'indication clinique. Pour les recommandations relatives à l'interruption du traitement, voir la rubrique «Posologie/Mode d'emploi».
Abus médicamenteux et dépendance au médicament
Aucune étude n'a été menée sur les abus médicamenteux ou la dépendance au médicament. Toutefois, les données disponibles n'indiquent pas que le traitement par Enspryng entraîne une dépendance.
La prudence est recommandée lors du passage à une autre forme galénique et/ou à un autre médicament contenant le même principe actif. Le patient doit être contrôlé de manière adéquate.
Tumeurs malignes
Les médicaments immunomodulateurs peuvent augmenter le risque de tumeurs malignes.
Les effets du traitement par Enspryng sur le développement d'affections malignes ne sont pas connus.
Réactions d'hypersensibilité
Des réactions d'hypersensibilité peuvent survenir chez les patients traités par des agents biologiques. Il convient d'informer les patients des éventuels symptômes d'une réaction d'hypersensibilité avant l'instauration du traitement.
La première injection doit être faite sous la surveillance d'un professionnel de la santé qualifié.
En cas d'apparition de réactions d'hypersensibilité, les patients doivent immédiatement en informer leur médecin et, le cas échéant, le consulter en urgence.
Événements cardiovasculaires
Les inhibiteurs du récepteur de l'IL-6 peuvent augmenter le risque de maladies cardiovasculaires. Une surveillance régulière est donc nécessaire, en particulier chez les patients ayant des facteurs de risque tels qu'hypertension artérielle, dyslipidémie et diabète (ECG, mesure de la pression artérielle).
Paramètres lipidiques
Des élévations des triglycérides et du cholestérol (paramètres lipidiques) ont été observées chez des patients traités par Enspryng (voir «Effets indésirables»). Les patients présentant une élévation des paramètres lipidiques doivent être traités conformément aux directives cliniques actuelles relatives au traitement de l'hyperlipidémie.
Activation du système du complément
Une activation du système du complément peut se produire lors d'un traitement par des inhibiteurs du récepteur de l'IL-6. Les données cliniques actuelles n'ont pas montré un tel effet sous Enspryng. Toutefois, les données étant limitées, le risque ne peut pas être évalué avec certitude pour Enspryng.
Affections démyélinisantes du SNC
La survenue d'autres affections inflammatoires du SNC a été observée lors d'un traitement par des inhibiteurs du récepteur de l'IL-6. Il convient d'être attentif aux symptômes évocateurs d'une affection démyélinisante du SNC d'apparition récente. Toutefois, les données étant limitées, le risque ne peut pas être évalué avec certitude pour Enspryng.
Perforation diverticulaire/'intestinale
La survenue de perforations diverticulaires/intestinales a été observée lors d'un traitement par des inhibiteurs du récepteur de l'IL-6 chez des patients atteints de polyarthrite rhumatoïde (PR). Les patients présentant des antécédents connus de diverticulite ont été exclus des études pivots menées avec Enspryng. Un risque accru de perforation diverticulaire/intestinale, comme observé avec d'autres inhibiteurs du récepteur de l'IL-6, ne peut pas être exclu au cours du traitement par Enspryng. Enspryng doit être utilisé avec prudence chez les patients présentant des antécédents d'ulcères intestinaux ou de diverticulite. En cas d'apparition de douleurs abdominales aiguës, les patients doivent être examinés sans délai afin de pouvoir dépister précocement une perforation gastro-intestinale.
Interactions
Aucune étude spécifique d'interactions n'a été réalisée avec Enspryng.
Interactions pharmacodynamiques
L'expression des enzymes hépatiques du CYP450 est réprimée par les cytokines qui stimulent les inflammations chroniques, telles que l'IL-6. Par conséquent, l'expression du CYP450 pourrait être modifiée lors du traitement par Enspryng entraînant une inhibition des cytokines.
Les patients prenant des médicaments dont la dose est ajustée individuellement et qui sont métabolisés par les CYP450 3A4, 1A2 ou 2C9 (p.ex. atorvastatine, inhibiteurs des canaux calciques, théophylline, warfarine, phénytoïne, cyclosporine ou benzodiazépines) doivent donc être contrôlés au début et à la fin d'un traitement par Enspryng et la dose de ces substances doit être ajustée si nécessaire. Compte tenu de la longue demi-vie d'élimination, l'effet d'Enspryng sur l'activité des enzymes du CYP450 peut persister pendant plusieurs semaines après l'arrêt du traitement.
Dans les analyses pharmacocinétiques de population, aucun effet de l'AZA, des corticostéroïdes ou du MMF n'a été observé sur la clairance d'Enspryng.
Grossesse/Allaitement
Grossesse
Il n'existe pas de données concernant l'emploi d'Enspryng chez la femme enceinte.
Les expérimentations chez le singe n'ont montré aucune toxicité directe ayant une incidence sur la grossesse, le développement fœtal et le développement postnatal. Chez l'animal, le satralizumab traverse la barrière placentaire et dans la progéniture d'animaux traités par le satralizumab, quelques observations ont éventuellement été en rapport avec l'effet pharmacologique sur l'IL-6 (voir la rubrique «Données précliniques, Toxicité sur la reproduction»). L'importance clinique de ces observations n'est pas connue.
Enspryng ne doit pas être administré pendant la grossesse, à moins que le bénéfice potentiel pour la mère prédomine sur le risque encouru par le fœtus. Le satralizumab traversant la barrière placentaire, les enfants nés de femmes traitées par Enspryng peuvent présenter un risque infectieux accru; la prudence est recommandée lors de l'administration de vaccins vivants à ces enfants.
Chez les femmes en âge de procréer, l'utilisation d'une méthode de contraception appropriée doit être envisagée pendant le traitement et jusqu'à 5 mois après la dernière dose d'Enspryng.
Allaitement
On ignore si Enspryng est excrété dans le lait maternel chez la femme ou est absorbé par voie systémique après l'absorption. Mais comme les IgG sont excrétées dans le lait maternel chez la femme et que l'excrétion dans le lait maternel a été démontrée dans des études précliniques (voir la rubrique «Données précliniques»), les femmes traitées par Enspryng ne doivent pas allaiter. Le bénéfice de l'allaitement pour l'enfant doit être évalué au regard du bénéfice du traitement pour la mère.
Fertilité
On ne dispose d'aucune donnée clinique sur les effets d'Enspryng sur la fertilité chez l'être humain. Les expérimentations animales ne suggèrent aucune altération de la fertilité masculine ou féminine (voir la rubrique «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machines
Aucune étude n'a été effectuée sur les effets sur l'aptitude à la conduite et l'utilisation de machines. Cependant, les données disponibles ne mettent en évidence aucune influence du traitement par Enspryng sur l'aptitude à la conduite et l'utilisation de machines.
Effets indésirables
Résumé du profil de sécurité
La sécurité d'Enspryng en monothérapie ou en association avec un TIS a été évaluée à l'aide des données de deux études cliniques de phase III, randomisées, multicentriques, en double aveugle et contrôlées contre placebo (BN40898 et BN40900), dans lesquelles 63 patients ont reçu Enspryng en monothérapie et 41 patients ont reçu Enspryng en association avec un TIS (voir la rubrique «Propriétés/Effets, Efficacité clinique»).
Les effets indésirables (EI) les plus fréquemment rapportés étaient des céphalées, des arthralgies, une leucopénie et des réactions liées à l'injection.
Les données de sécurité disponibles concernant le nombre de patients exposés à Enspryng ainsi que la durée de l'exposition sont limitées. Il se peut que des EI potentiels, relativement rares et éventuellement graves, passent inaperçus dans le programme d'études. D'éventuels effets de classe observés avec d'autres inhibiteurs du récepteur de l'IL-6 tels que des perforations intestinales, des infections opportunistes dont la tuberculose, une réactivation de l'hépatite B, une augmentation du risque cardiovasculaire, une activation du système du complément, des affections démyélinisantes, ainsi que des tumeurs malignes ne sont pas exclus (voir la rubrique «Mises en garde et précautions»).
Tableau récapitulatif des effets indésirables issus des études cliniques
Les EI rapportés au cours des études cliniques en lien avec l'utilisation d'Enspryng en monothérapie ou en association avec un TIS sont résumés dans le Tableau 1. Dans les deux études cliniques, les patients des groupes Enspryng ont été traités plus longtemps que les patients du groupe placebo (ou du groupe ayant utilisé le placebo en association avec un TIS). Les EI issus des études cliniques (Tableau 1) sont répertoriés selon les classes de systèmes d'organes MedDRA. La catégorie de fréquence pour chaque effet indésirable se base sur la convention suivante: très fréquents (≥1/10), fréquents (≥1/100 à <1/10), occasionnels (≥1/1'000 à <1/100), rares (≥1/10'000 à <1/1'000), très rares (<1/10'000).
Tableau 1: Résumé des EI survenus chez les patients traités par Enspryng en monothérapie ou en association avec un traitement immunosuppresseur au cours des études cliniques
Effets indésirables (MedDRA) | Nombre de patients (%) | Catégorie de fréquence pour Enspryng | |
Enspryng | Placebo1 | ||
Infections et infestations | |||
Infections des voies urinaires | 18 (17,3%) | 15 (20,3%) | Très fréquents |
Infections des voies respiratoires supérieures | 20 (19,2%) | 12 (16,2%) | Très fréquents |
Rhinopharyngite | 19 (18,3%) | 8 (10,8%) | Très fréquents |
Grippe (influenza) | 5 (4,8%) | 6 (8,1%) | Fréquents |
Affections du système nerveux | |||
Céphalées | 20 (19,2%) | 8 (10,8%) | Très fréquents |
Migraine | 4 (3,8%) | 0 | Fréquents |
Lésions, intoxications et complications liées aux procédures | |||
Réactions liées à l'injection | 13 (12,5%) | 7 (9,5%) | Très fréquents |
Affections musculosquelettiques et du tissu conjonctif | |||
Arthralgie | 14 (13,5%) | 1 (1,4%) | Très fréquents |
Raideur de l'appareil locomoteur | 5 (4,8%) | 0 | Fréquents |
Affections de la peau et du tissu sous-cutané | |||
Éruption cutanée | 9 (8,7%) | 3 (4,1%) | Fréquents |
Prurit | 6 (5,8%) | 1 (1,4%) | Fréquents |
Affections psychiatriques | |||
Insomnie | 6 (5,8%) | 1 (1,4%) | Fréquents |
Troubles généraux et anomalies au site d'administration | |||
Œdème, périphérique | 5 (4,8%) | 0 | Fréquents |
Affections hématologiques et du système lymphatique | |||
Hypofibrinogénémie | 3 (2,9%) | 0 | Fréquents |
Affections respiratoires, thoraciques et médiastinales | |||
Rhinite, allergique | 4 (3,8%) | 0 | Fréquents |
Troubles du métabolisme et de la nutrition | |||
Hyperlipidémie | 14 (13,5%) | 9 (12,2%) | Très fréquents |
Investigations | |||
Leucopénie | 14 (13,5%) | 4 (5,4) | Très fréquents |
Hyperbilirubinémie | 2 (1,9%) | 0 | Fréquents |
Augmentation du poids corporel | 5 (4,8%) | 2 (2,7%) | Fréquents |
Thrombopénie | 4 (3,8%) | 2 (2,7%) | Fréquents |
Augmentation des transaminases | 7 (6,7%) | 6 (8,1%) | Fréquents |
1 Placebo ou placebo en association avec un TIS
Description de certains effets indésirables
Réactions liées à l'injection (IRR)
Les IRR rapportées chez les patients traités par Enspryng en monothérapie ou en association avec un TIS étaient en majorité légères à modérées et sont survenues le plus souvent dans les 24 heures ayant suivi les injections. Les symptômes systémiques les plus fréquemment rapportés étaient une diarrhée et des céphalées. Les réactions locales au site d'injection les plus fréquemment rapportées étaient une sensation de chaleur, un érythème, un prurit, une éruption cutanée et des douleurs. Aucune des réactions liées à l'injection n'a nécessité l'interruption ou l'arrêt du traitement.
Infections
Afin de tenir compte de la durée plus longue du traitement dans le groupe satralizumab par rapport au groupe placebo, les données sur les infections ne sont pas seulement données en pourcentage, mais également en événements par 100 années-patients (événements survenant chez 100 patients après un an de traitement).
Dans l'étude avec Enspryng en monothérapie, la fréquence des infections chez les patients traités avec Enspryng était de 99,8 événements/100 années-patients (IC à 95%: 82,4; 119,8) ou 34/63 patients [54,0% (IC à 95%: 40,94%; 66,61%)] contre 162,6 événements/100 années-patients (IC à 95%: 125,8; 206,9) ou 14/32 patients [43,8% (IC à 95%: 26,36%; 62,34%)] chez les patients du groupe placebo. La fréquence des infections graves était de 5,2 événements/100 années-patients (IC à 95%: 1,9; 11,3) ou 6/63 patients [9,5% (IC à 95%: 3,58%; 19,59%)] chez les patients traités par Enspryng contre 9,9 événements/100 années-patients (IC à 95%: 2,7; 25,2) ou 3/32 patients [9,4% (IC à 95%: 1,98%; 25,02%)] chez les patients du groupe placebo.
Chez les patients traités par Enspryng en association avec un TIS, la fréquence des infections était de 132,5 événements/100 années-patients (IC à 95%: 108,2; 160,5) ou 28/41 patients [68,3% (IC à 95%: 51,91%; 81,92%)] contre 149,6 événements/100 années-patients (IC à 95%: 120,1; 184,1) ou 26/42 patients [61,9% (IC à 95%: 45,64%; 76,43%)] chez les patients ayant reçu le placebo en association avec un TIS. La fréquence des infections graves était de 2,6 événements/100 années-patients (IC à 95%: 0,3; 9,2) ou 2/41 patients [4,9% (IC à 95%: 0,60%; 16,53%)] contre 5,0 événements/100 années-patients (IC à 95%: 1,0; 14,7) ou 3/42 patients [7,1% (IC à 95%: 1,50%; 19,48%)] chez les patients ayant reçu le placebo en association avec un TIS.
Augmentation du poids corporel
Durant la période de traitement en double aveugle, une augmentation du poids corporel d'au moins 15% par rapport à la valeur initiale a été observée chez 4,8% des patients traités par Enspryng (en monothérapie ou en association avec un traitement immunosuppresseur) et chez 2,7% des patients ayant reçu le placebo (ou le placebo en association avec un traitement immunosuppresseur).
Anomalies biologiques
Neutrophiles
Durant la période de traitement en double aveugle, une diminution du nombre de neutrophiles a été observée chez 31,7% des patients traités par Enspryng (en monothérapie ou en association avec un TIS) et chez 21,6% des patients ayant reçu le placebo (seul ou en association avec un TIS). La diminution du nombre de neutrophiles a le plus souvent été passagère ou intermittente.
Parmi les patients du bras Enspryng, 9,6% avaient un taux de neutrophiles inférieur à 1 x 109/l versus 5,4% dans le bras placebo ou dans le bras placebo plus TIS, ce qui n'a pas été associé temporellement à des infections graves.
Thrombocytes
Durant la période de traitement en double aveugle, une diminution du nombre de thrombocytes a été observée chez 24,0% des patients traités par Enspryng (en monothérapie ou en association avec un TIS) et chez 9,5% des patients ayant reçu le placebo (seul ou en association avec un TIS). Ces diminutions du nombre de thrombocytes n'ont pas été associées à des événements hémorragiques.
La diminution du nombre de thrombocytes a été passagère et n'était pas inférieure à 75 x 109/l. Chez aucun patient, le taux de thrombocytes n'a été ≤50 x 109/l.
Enzymes hépatiques
Durant la période de traitement en double aveugle, des élévations des taux d'ALAT ou d'ASAT ont été observées chez respectivement 27,9% et 18,3% des patients traités par Enspryng (en monothérapie ou en association avec un TIS) et chez respectivement 12,2% et 13,5% des patients ayant reçu le placebo (seul ou en association avec un TIS). Les élévations ont le plus souvent été inférieures à 3 fois la LSN et ont été passagères et réversibles sans interruption du traitement par Enspryng.
Des élévations des taux d'ALAT ou d'ASAT >3 x LSN sont survenues chez respectivement 2,9% et 1,9% des patients traités par Enspryng en monothérapie et en association avec un TIS, mais n'ont pas été associées à une augmentation de la bilirubine totale. Chez un patient ayant reçu Enspryng en association avec un TIS, des élévations des taux d'ALAT >5 x LSN ont été observées 4 semaines après l'instauration du traitement et les taux d'ALAT se sont normalisés après l'arrêt du traitement par Enspryng.
Paramètres lipidiques
Durant la période de traitement en double aveugle, une élévation du cholestérol total supérieure à 7,75 mmol/l a été observée chez 10,6% des patients traités par Enspryng (en monothérapie ou en association avec un TIS) et chez 1,4% des patients ayant reçu le placebo (seul ou en association avec un TIS). Des élévations des triglycérides supérieures à 3,42 mmol/l sont survenues chez 18,3% des patients traités par Enspryng versus 6,8% des patients sous placebo. Les élévations des paramètres lipidiques n'ont pas nécessité d'interruption du traitement.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
Aucun cas de surdosage n'a été rapporté chez des patients atteints de NMO ou de NMOSD. Aucune donnée n'est disponible concernant des surdosages chez les patients atteints de NMO ou de NMOSD.
Signes et symptômes
Dans une étude de phase I, des volontaires adultes sains ont reçu par voie sous-cutanée une dose unique d'Enspryng allant jusqu'à 240 mg, sans que des effets indésirables graves ou sévères ne soient survenus.
Traitement
En cas de surdosage, le patient doit être étroitement surveillé et doit recevoir un traitement symptomatique. Des mesures de soutien doivent être instaurées si nécessaire.
Propriétés/Effets
Code ATC:
L04AC19
Mécanisme d'action
Le satralizumab est un anticorps monoclonal (mAk) humanisé de type IgG2 qui se lie aux récepteurs solubles et membranaires de l'IL-6 humaine (IL-6R) et inhibe ainsi la transmission du signal médié par ces récepteurs de l'IL-6.
L'IL-6 est une cytokine pléiotrope produite par un grand nombre de types cellulaires et impliquée dans différents processus inflammatoires, tels que l'activation des lymphocytes B, la différenciation des lymphocytes B en plasmocytes et la production d'auto-anticorps, l'activation et la différenciation des lymphocytes Th17, l'inhibition des lymphocytes T régulateurs et les modifications de la perméabilité de la barrière hémato-encéphalique. Les taux d'IL-6 dans le liquide cérébrospinal et le sérum de patients atteints de NMO et de NMOSD sont augmentés pendant les phases où la maladie est active. Certaines fonctions de l'IL-6 sont mises en relation avec la pathogenèse de la NMO et de la NMOSD, dont la formation d'auto-anticorps pathologiques dirigés contre l'aquaporine-4 (AQP4), une protéine du canal hydrique exprimée essentiellement par les astrocytes dans le SNC.
Pharmacodynamique
Les études cliniques menées avec Enspryng dans la NMO et la NMOSD ont montré des diminutions des taux de protéine C réactive (CRP), de fibrinogène et de complément (C3, C4 et CH50).
Efficacité clinique
L'efficacité et la sécurité d'Enspryng ont été évaluées dans deux études cliniques pivots de phase III (BN40898 et BN40900), menées chez des patients porteurs d'un diagnostic de NMO séropositive ou séronégative pour les IgG anti-AQP4 (critères de Wingerchuk de 2006) ou porteurs d'un diagnostic de NMOSD séropositive pour les IgG anti-AQP4 (critère de Wingerchuk de 2007). Rétrospectivement, ces patients remplissaient également les tout récents critères proposés par le comité international pour le diagnostic de la NMO (Réf: Wingerchuk et al. 2015). Les effets d'Enspryng ont été évalués chez des patients adultes (dans les études BN40898 et BN40900) et chez au total N=7 patients adolescents âgés de ≥12 à <18 ans (N=4 Enspryng, N=3 placebo) (dans l'étude BN40898). Dans les deux études, l'inclusion de patients adultes atteints de NMO et séronégatifs pour les IgG anti-AQP4 était limitée à environ 30% afin que la population de l'étude reflète la population réelle de patients atteints de NMO.
Dans les deux études, l'indicateur primaire de l'efficacité était le nombre de rechutes définies par protocole (protocol-defined relapse, PDR) sur la base d'une aggravation prédéfinie des scores EDSS (Expanded Disability Status Scale) et FSS (Functional System Scores), et confirmées par un CEC (Clinical Endpoint Committee) indépendant. Le principal critère d'évaluation pour l'analyse était le délai jusqu'à la première PDR confirmée par le CEC, avec évaluation des scores EDSS/FSS dans les 7 jours suivant la déclaration des symptômes par le patient (rechute évaluée).
Étude BN40898 (SA-307JG ou SAkuraSky)
L'étude clinique BN40898 randomisée, multicentrique, en double aveugle et contrôlée contre placebo visait à évaluer l'efficacité d'Enspryng en association avec un TIS stable (jusqu'à 15 mg/jour de CSO [équivalent de prednisolone], jusqu'à 3 mg/kg/jour d'AZA ou jusqu'à 3'000 mg/jour de MMF; les adolescents ont reçu une association d'AZA et de CSO, ou de MMF et de CSO). 83 patients séropositifs et séronégatifs pour les IgG anti-l'AQP4 (dont 7 adolescents) ont participé à cette étude. Les patients ont reçu les 3 premières doses unitaires de 120 mg d'Enspryng ou un placebo correspondant en injection sous-cutanée dans la paroi abdominale ou dans la cuisse, toutes les 2 semaines pendant les 4 premières semaines, puis une fois toutes les 4 semaines.
La conception de l'étude et les caractéristiques initiales de la population de l'étude figurent dans le Tableau 2. L'étude était pilotée par les événements et la phase en double aveugle de l'étude visant à évaluer l'efficacité s'est achevée après l'apparition de 26 rechutes évaluées. Les patients qui avaient présenté une PDR confirmée par le CEC pendant la phase en double aveugle (DA), qui avaient reçu un traitement de secours en raison d'une rechute ou qui avaient terminé la phase en DA, pouvaient être inclus dans la phase d'extension ouverte (open-label extension – OLE) durant laquelle tous les patients ont été traités en ouvert par Enspryng.
Tableau 2: Conception de l'étude et caractéristiques initiales de l'étude BN40898
Nom de l'étude | Étude BN40898 (N=83) | |
Conception de l'étude | ||
Population de l'étude | Patients adolescents et adultes atteints de NMO ou de NMOSD recevant un traitement stable par un TIS Âge de 12-74 ans, ≥2 rechutes dans les 2 ans précédant l'inclusion (avec au moins une rechute dans les 12 mois précédant l'inclusion), EDSS de 0 à 6,5 | |
Durée de l'étude en vue de l'évaluation de l'efficacité | Pilotée par les événements (26 rechutes définies par protocole et confirmées par le CEC) Durée médiane du suivi: Enspryng 100 semaines, placebo 74 semaines | |
Groupes de traitement, randomisation 1:1 | Groupe A: 120 mg d'Enspryng s. c. Groupe B: placebo | |
Caractéristiques initiales | Enspryng + TIS (n = 41) | Placebo + TIS (n = 42) |
Diagnostic, n (%): |
|
|
NMO NMOSD | 33 (80,5) 8 (19,5) | 28 (66,7) 14 (33,3) |
Séropositivité des IgG anti-AQP4, n (%) | 27 (65,9) | 28 (66,7) |
Âge moyen an années (écart-type) | 40,8 (16,1) (13 – 73) | 43,4 (12,0) (14 – 65) |
Adolescents (≥12 et <18 ans), n (%) | 4 (9,8) | 3 (7,1) |
Répartition selon le sexe, |
|
|
n (%) sexe masculin/ n (%) sexe féminin | 4 (9,8) / 37 (90,2) | 2 (4,8) / 40 (95,2) |
Traitement immunosuppresseur (TIS), n (%): |
|
|
corticostéroïdes oraux (CSO)) azathioprine (AZA) mycophénolate mofétil (MMF) AZA + CSO* MMF + CSO* | 17 (41,5) 16 (39,0) 4 (9,8) 3 (7,3) 1 (2,4) | 20 (47,6) 13 (31,0) 8 (19,0) 0 1 (2,4) |
* Association autorisée pour les patients adolescents
Étude BN40900 (SA-309JG ou SAkuraStar)
L'étude clinique BN40900 randomisée, multicentrique, en double aveugle et contrôlée contre placebo visait à évaluer l'efficacité d'Enspryng en monothérapie par rapport à un placebo. 95 patients adultes séropositifs et séronégatifs pour les IgG anti-AQP4 ont participé à cette étude. Les patients ont reçu les 3 premières doses unitaires de 120 mg d'Enspryng ou un placebo correspondant, en injection sous-cutanée dans la paroi abdominale ou dans la cuisse, toutes les 2 semaines pendant les 4 premières semaines, puis une fois toutes les 4 semaines.
La conception de l'étude et les caractéristiques initiales de la population de l'étude figurent dans le Tableau 3.
La phase en double aveugle de l'étude visant à évaluer l'efficacité s'est achevée 1,5 an après la date de randomisation du dernier patient admis. Les patients qui avaient présenté une PDR confirmée par le CEC pendant la phase en DA ou qui avaient terminé la phase en DA, pouvaient être inclus dans la phase OLE, durant laquelle tous les patients ont été traités en ouvert par Enspryng.
Tableau 3: Conception de l'étude et caractéristiques initiales de l'étude BN40900
Nom de l'étude | Étude BN40900 (N = 95) | |
Conception de l'étude | ||
Population de l'étude | Patients adultes atteints de NMO ou de NMOSD Âge de 18-74 ans, ≥1 rechute ou première poussée dans les 12 mois précédant l'inclusion, EDSS de 0 à 6,5. Les patients avaient reçu un traitement antérieur pour prévenir une rechute de NMOSD ou n'avaient encore jamais reçu de traitement. | |
Durée de l'étude en vue de l'évaluation de l'efficacité | Pilotée par les événements (44 rechutes définies par protocole, confirmées par le CEC ou 1,5 an après la date de randomisation du dernier patient admis, selon ce qui survient en premier) Durée médiane du suivi: Enspryng 95,4 semaines, placebo 60,5 semaines | |
Groupes de traitement, randomisation 2:1 | Monothérapie: Groupe A: 120 mg d'Enspryng s. c. Groupe B: placebo | |
Caractéristiques initiales | Enspryng (n = 63) | Placebo (n = 32) |
Diagnostic, n (%): |
|
|
NMO NMOSD | 47 (74,6) 16 (25,4) | 24 (75,0) 8 (25,0) |
Séropositivité des IgG anti-AQP4, n (%) | 41 (65,1) | 23 (71,9) |
Âge moyen an années (écart-type) | 45,3 (12,0) (21 – 70) | 40,5 (10,5) (20 – 56) |
Répartition selon le sexe, |
|
|
n (%) sexe masculin/ n (%) sexe féminin | 17 (27,0) / 46 (73,0) | 1 (3,1) / 31 (96,9) |
Efficacité primaire - phase en double aveugle
Dans la population en ITT, le traitement par Enspryng a entraîné une réduction statistiquement significative, de 62% (hazard ratio [HR] [IC à 95%]: 0,38 [0,16-0,88]; p [test du Log-Rang] = 0,0184), du risque d'apparition d'une rechute évaluée par rapport au placebo lors de l'utilisation en association avec un TIS stable (étude BN40898), et une réduction de 55% (HR [IC à 95%]: 0,45 [0,23-0,89]; p [test du Log-Rang] = 0,0184) du risque d'apparition d'une rechute évaluée par rapport au placebo lors de l'utilisation en monothérapie (étude BN40900).
L'effet de sous-groupe le plus important a été observé chez les patients séropositifs pour les IgG anti-AQP4. Chez les patients séropositifs pour les IgG anti-AQP4, le risque relatif d'apparition d'une rechute évaluée était réduit de 79% dans l'étude BN40898 (HR [IC à 95%]: 0,21 [0,06-0,75]) et de 74% dans l'étude BN40900 (HR [IC à 95%]: 0,26 [0,11-0,63]). À 48 semaines, respectivement 91,5% et 82,9% des patients séropositifs pour les IgG anti-AQP4 traités par Enspryng en association avec un TIS et en monothérapie n'avaient présenté aucune rechute évaluée. À 96 semaines, respectivement 91,5% et 76,5% des patients séropositifs pour les IgG anti-AQP4 traités par Enspryng en association avec un TIS et en monothérapie n'avaient présenté aucune rechute évaluée (voir le Tableau 4 et la Fig. 1 et 2). Lorsque les données des études BN40898 et BN40900 ont été combinées, le traitement par Enspryng avec ou sans TIS a entraîné une réduction totale du risque de 75% chez les patients séropositifs pour les IgG anti-AQP4 (HR [IC à 95%]; 0,25 [0,12-0,50]). Les différences concernant le délai jusqu'à la première rechute évaluée dans le groupe des patients séronégatifs pour les IgG anti-AQP4 n'étaient pas significatives entre les patients qui avaient reçu Enspryng avec ou sans TIS et les patients qui avaient reçu le placebo avec ou sans TIS (BN40898 et BN40900 combinées: HR [IC à 95%]: 0,97 [0,41-2,33]).
Tableau 4: Principaux critères d'efficacité des études BN40898 et BN40900 chez les patients séropositifs pour les IgG anti-AQP4
BN40898 | BN40900 | |||
Enspryng | Placebo | Enspryng | Placebo | |
Principal critère d'évaluation chez les patients séropositifs pour les IgG anti-AQP4 | ||||
Nombre de patients séropositifs pour les IgG anti-AQP4 (n) | 27 | 28 | 41 | 23 |
Réduction du risque (études individuelles) | 79% (HR: 0,21; IC à 95%: 0,06; 0,75; p = 0,0086) | 74% (HR: 0,26; IC à 95%: 0,11; 0,63; p = 0,0014) | ||
Réduction du risque (analyse groupée) | 75% (HR: 0,25; IC à 95%: 0,12; 0,50; p: <0,0001) | |||
Pourcentage de patients sans rechute évaluée à 48 semaines | 91,5% (IC à 95%: 69,64; 97,83) | 59,9% (IC à 95%: 36,25; 77,25) | 82,9% (IC à 95%: 67,49; 91,47) | 55,4% (IC à 95%: 32,96; 73,08) |
Pourcentage de patients sans rechute évaluée à 96 semaines | 91,5% (IC à 95%: 69,64; 97,83) | 53,3% (IC à 95%: 29,34; 72,38) | 76,5% (IC à 95%: 59,22; 87,21) | 41,1% (IC à 95%: 20,76; 60,41) |
Fig. 1: Étude BN40898: délai jusqu'à la première rechute évaluée pendant la phase en double aveugle chez les patients séropositifs pour les IgG anti-AQP4

Fig. 2: Étude BN40900: délai jusqu'à la première rechute évaluée pendant la phase en double aveugle chez les patients séropositifs pour les IgG anti-AQP4

Le traitement par Enspryng des patients séropositifs pour les IgG anti-AQP4 a réduit le taux annualisé de rechutes évaluées (Adjudicated Relapse Rate – ARR) de 88% dans l'étude BN40898 et de 90% dans l'étude BN40900 (Tableau 5) par rapport au placebo.
Tableau 5: Taux annualisé de rechutes évaluées pendant la phase en double aveugle (modèle de régression binomiale négative)
BN40898 | BN40900 | Groupées | |||||
Placebo | Enspryng | Placebo | Enspryng | Placebo | Enspryng | ||
Patients séropositifs pour les IgG anti-AQP4 | N = 28 | N = 27 | N = 23 | N = 41 | N = 51 | N = 68 | |
Patients avec rechute | 12 | 3 | 13 | 9 | 25 | 12 | |
Taux annualisé ajusté de rechutes | 0,520 | 0,063 | 2,853 | 0,275 | 1,339 | 0,136 | |
Réduction relative de l'ARR (rapport de taux) | 88% (RR: 0,122; IC à 95%: 0,027; 0,546; p=0,0039) | 90% (RR: 0,096; IC à 95%: 0,020; 0,473; p= 0,0086) | 90% (RR: 0,102; IC à 95%: 0,034; 0,301; p=0,0002) | ||||
La nécessité de recourir à un traitement de secours (p.ex. corticostéroïdes, immunoglobulines intraveineuses et/ou aphérèse [incluant plasmaphérèse et échange plasmatique]) a été plus rare chez les patients séropositifs pour les IgG anti-AQP4 traités par Enspryng que chez les patients ayant reçu le placebo, et ce, de 61% dans l'étude BN40898 et de 74% dans l'étude BN40900.
Tableau 6: Recours à un traitement de secours chez les patients ayant présenté une rechute pendant la phase en double aveugle
BN40898 | BN40900 | Groupées | ||||
Placebo | Enspryng | Placebo | Enspryng | Placebo | Enspryng | |
Patients séropositifs pour les IgG anti-AQP4 | N = 28 | N = 27 | N = 23 | N = 41 | N = 51 | N = 68 |
Patients avec un traitement de secours | 18 (64,29%) | 11 (40,74%) | 14 (60,87%) | 13 (31,71%) | 32 (62,75%) | 24 (35,29%) |
Réduction du risque (Odds ratio) | 61% (OR: 0,3930; IC à 95%: 0,1343; 1,1502; p=0,0883) | 74% (OR: 0,2617; IC à 95%: 0,0862; 0,7943; p=0,0180) | 66% (OR: 0,3430; IC à 95%: 0,1614; 0,7289; p=0,0054) | |||
Le traitement par Enspryng de patients séropositifs pour les IgG anti-AQP4 a réduit le risque de rechute sévère (définie par une augmentation d'au moins 2 points d'EDSS par rapport au score EDSS antérieur) de 85% dans l'étude BN40898 et de 79% dans l'étude BN40900 (Tableau 7) par rapport au placebo.
Tableau 7: Délai jusqu'à la première rechute évaluée pendant la phase en double aveugle
BN40898 | BN40900 | Groupées | ||||
Placebo | Enspryng | Placebo | Enspryng | Placebo | Enspryng | |
Patients séropositifs pour les IgG anti-AQP4 | N=27 | N=27 | N=23 | N=41 | N=50 | N=68 |
Patients avec un événement | 6 (22,2%) | 1 (3,7%) | 5 (21,7%) | 3 (7,3%) | 11 (22,0%) | 4 (5,9%) |
Réduction du risque | 85% (HR: 0,15; IC à 95%: 0,02; 1,25; p=0,0441) | 79% (HR: 0,21; IC à 95%: 0,05; 0,91; p=0,0231) | 82% (HR: 0,18; IC à 95%: 0,06; 0,58; p=0,0015) | |||
Le traitement par Enspryng de patients séropositifs pour les IgG anti-AQP4 a réduit le risque d'aggravation du score EDSS par rapport au score initial de 65% dans l'étude BN40898 et de 66% dans l'étude BN40900 par rapport au placebo (Tableau 8).
Tableau 8: Délai d'aggravation du score sur l'échelle d'évaluation du handicap (Expanded Disability Status Scale, EDSS)* durant la phase en double aveugle (patients séropositifs pour les IgG anti-AQP4)
BN40898 | BN40900 | Combinées | |||||
Placebo | Enspryng | Placebo | Enspryng | Placebo | Enspryng | ||
Patients séropositifs pour les IgG anti-AQP4 | N = 27 | N = 28 | N = 23 | N = 41 | N = 50 | N = 69 | |
Patients avec un événement | 12 (44,4%) | 5 (17,9%) | 11 (47,8%) | 11 (26,8%) | 23 (46,0%) | 16 (23,2%) | |
Réduction du risque | 65% (HR: 0,35; IC à 95%: 0,12; 1,00; p = 0,0407) | 66% (HR: 0,34; IC à 95%: 0,14; 0,82; p = 0,0124) | 69% (HR: 0,31; IC à 95%: 0,16; 0,59; p = 0,0002) | ||||
* Une aggravation du score EDSS par rapport au score initial était définie par une: a) aggravation du score EDSS d'au moins 2 points chez les patients avec un score initial de 0, b) aggravation du score EDSS d'au moins 1 point chez les patients avec un score initial compris entre 1 et 5, c) aggravation du score EDSS d'au moins 0,5 point chez les patients avec un score initial d'au moins 5,5.
Phase d'extension ouverte
Des analyses de données à long terme, dont celles de la phase d'extension ouverte en cours (reposant sur les rechutes ayant été traitées par un traitement de secours), ont montré que respectivement 58% et 73% des patients séropositifs pour les IgG anti-AQP4-IgG et traités par Enspryng n'avaient pas présenté de rechutes après un traitement de 120 semaines, lorsqu'Enspryng avait été administré en traitement adjuvant ou en monothérapie.
On ne dispose globalement que de données limitées sur l'efficacité et la sécurité du traitement par Enspryng au-delà de 2 ans.
Fig. 3: Étude BN40898: délai jusqu'à la première rechute (rechute clinique traitée) pendant la phase en double aveugle et la phase ouverte chez les patients séropositifs pour les IgG anti-AQP4

Fig. 4: Étude BN40900: délai jusqu'à la première rechute (rechute clinique traitée) pendant la phase en double aveugle et la phase ouverte chez les patients séropositifs pour les IgG anti-AQP4

Caractéristiques à l'inclusion et efficacité chez les patients adolescents (étude BN40898)
L'âge moyen des 7 patients adolescents inclus dans l'étude BN40898 pendant la phase en double aveugle était de 15,4 ans et le poids corporel médian était de 79,6 kg. La plupart de ces patients adolescents étaient de sexe féminin (n = 6). Quatre patients étaient des Blancs, 2 patients étaient des Noirs/Afro-américains et 1 patient était asiatique. À l'inclusion, 3 des 7 (42,9%) patients adolescents étaient séropositifs pour les IgG anti-AQP4 (2 dans le groupe placebo et 1 dans le groupe Enspryng). Une rechute évaluée est survenue pendant la phase en double aveugle chez 1 des 3 adolescents du groupe placebo et chez 1 des 4 adolescents du groupe Enspryng. En raison du faible nombre de cas, le hazard ratio du critère principal, le délai jusqu'à la première rechute évaluée, n'a pas été calculé dans ce sous-groupe.
Informations complémentaires
Immunogénicité
Dans l'étude de phase III BN40898 (association avec un TIS) et dans l'étude de phase III BN40900 (monothérapie), des anticorps anti-médicament (AAM) ont été détectés chez respectivement 41% et 71% des patients ayant reçu Enspryng pendant la phase en double aveugle. La capacité de ces AAM à neutraliser la liaison d'Enspryng à sa cible est inconnue.
L'exposition était plus faible chez les patients AAM-positifs, mais les AAM n'ont eu aucun effet sur la sécurité ni aucun effet sur l'efficacité ou les marqueurs pharmacodynamiques de liaison aux molécules cibles.
Dans les études de phase III, le traitement par le satralizumab a entraîné des réductions similaires du risque de survenue d'une rechute évaluée chez les patients, bien que les fréquences des AAM aient été différentes selon les patients. Les patients ayant un poids corporel plus élevé et un taux d'exposition plus faible ont développé des AAM avec une plus grande probabilité (qu'ils aient reçu ou non un traitement de fond par un TIS), mais les effets thérapeutiques étaient comparables dans tous les groupes définis en fonction du poids, lorsque l'administration a eu lieu en association avec un TIS ou en monothérapie.
La dose recommandée convient à tous les patients et il n'est pas nécessaire d'interrompre le traitement ou de modifier la dose chez les patients développant des AAM.
Pharmacocinétique
La pharmacocinétique d'Enspryng a été décrite chez des volontaires sains japonais et caucasiens et chez des patients atteints de NMO et de NMOSD. Le profil pharmacocinétique chez les patients atteints de NMO et de NMOSD a été caractérisé lors de l'administration de la dose recommandée à l'aide d'analyses pharmacocinétiques de population.
L'évolution dans le temps de la concentration d'Enspryng chez des patients atteints de NMO ou de NMOSD a été décrite par un modèle PC de population à deux compartiments avec en parallèle une élimination linéaire et une élimination médiée par la cible (cinétique de Michaelis-Menten), et une absorption sous-cutanée (s. c.) de premier ordre. Les paramètres de clairance et de volume d'Enspryng ont été ajustés par allométrie en fonction du poids corporel (à l'aide d'une fonction puissance avec un coefficient de puissance fixe respectivement de 0,75 et 1 pour les paramètres de clairance et de volume). Le poids corporel s'est avéré être une covariable significative, la clairance et le Vc (volume de distribution central) ayant augmenté respectivement de 71,3% et de 105% chez les patients pesant 123 kg (97,5e percentile de la distribution du poids) par rapport à un patient pesant 60 kg.
L'état d'équilibre a été atteint après la phase de charge (8 semaines), avec les valeurs suivantes pour la Cmin, la Cmax et l'AUC (moyenne (± écart-type)): Cmin: 28,5 (10,2) µg/ml, Cmax: 42,7 (11,9) µg/ml et AUC: 1020 (313) µg.ml/jour. Le traitement immunologique de fond n'a pas influencé la pharmacocinétique (voir la rubrique «Interactions»).
Absorption
La constante de vitesse d'absorption d'Enspryng s'est élevée à 0,251 1/jour (IC à 95%: 0,216-0,285), ce qui correspond à une demi-vie d'absorption d'environ 3 jours lors de l'administration de la dose recommandée (voir la rubrique «Posologie/Mode d'emploi»). La biodisponibilité était élevée (85,4%, IC à 95%: 0,795-0,953).
Distribution
La distribution d'Enspryng est biphasique. Le volume de distribution central s'est élevé à 3,46 l (IC à 95%: 3,21-3,97) et le volume de distribution périphérique à 2,07 l (IC à 95%: 1,78-2,59). La clairance intercompartimentale a été de 0,336 l/jour (IC à 95%: 0,261-0,443).
Métabolisme
Le métabolisme d'Enspryng n'a pas été directement étudié. Enspryng devrait être éliminé par catabolisme.
Élimination
La clairance d'Enspryng est concentration-dépendante. La clairance linéaire est estimée à 0,0601 l/jour (IC à 95%: 0,0524-0,0695) et représente environ la moitié de la clairance totale à l'état d'équilibre chez les patients atteints de NMO et de NMOSD ayant reçu la dose recommandée. Sur la base des données combinées issues des études de phase III, la demi-vie terminale (t1/2) associée est d'environ 30 jours (intervalle: 22-37 jours).
Cinétique pour certains groupes de patients
Les analyses pharmacocinétiques de population réalisées chez des patients adultes atteints de NMO ou de NMOSD n'ont révélé aucun effet de l'âge, du sexe et de l'origine ethnique sur la pharmacocinétique du satralizumab. Même si le poids corporel a eu un impact sur la pharmacocinétique du satralizumab, aucun ajustement de la dose n'est recommandé pour tenir compte de ces caractéristiques démographiques.
Troubles de la fonction hépatique
Aucune étude spécifique n'a été effectuée sur les effets de l'insuffisance hépatique sur la PC du satralizumab.
Troubles de la fonction rénale
Aucune étude spécifique n'a été effectuée sur les effets de l'insuffisance rénale sur la PC du satralizumab. Toutefois, 22 patients présentant une insuffisance rénale légère (clairance de la créatinine <80 ml/min et ≥50 ml/min) ont été inclus dans les études cliniques BN40898 et BN40900. Comme les mécanismes connus d'élimination du satralizumab permettaient de s'y attendre, la PC n'a pas été pas modifiée chez ces patients et aucun ajustement posologique n'est donc nécessaire.
Patients âgés
Aucune étude spécifique n'a été réalisée pour évaluer la PC du satralizumab chez les patients de plus de 65 ans. Toutefois, des patients atteints de NMO ou de NMOSD âgés de 65 à 74 ans ont été inclus dans les études BN40898 et BN40900.
Les analyses pharmacocinétiques de population basées sur les données de ces patients ont montré que l'âge n'avait aucune influence sur la PC du satralizumab.
Enfants et adolescents
Les données de 8 patients adolescents [13-17 ans], chez lesquels le schéma posologique des adultes a été utilisé, montrent que les paramètres PC de population du satralizumab ne sont pas significativement différents de ceux des adultes.
Aucun ajustement posologique n'est donc nécessaire.
Données précliniques
Pharmacologie de sécurité/Toxicité à long terme (ou toxicité en cas d'administration répétée)
Des données précliniques chez le singe, une espèce pertinente affichant une réactivité croisée au satralizumab, n'ont pas révélé de risque particulier pour l'être humain sur la base de la pharmacologie de sécurité et de critères de toxicité. Aucun effet indésirable n'a été observé au cours des études de toxicité de 4 et 26 semaines, lors de l'administration sous-cutanée répétée de doses maximales de 50 mg de satralizumab par kg une fois par semaine à des singes cynomolgus. La seule modification pertinente observée dans ces études était une augmentation du taux sanguin d'IL-6, considérée comme le résultat de l'effet pharmacologique (blocage du IL-6R) du satralizumab. Chez la plupart des animaux traités, le traitement par le satralizumab a induit une réponse immunitaire avec des anticorps anti-médicament, ce qui n'a cependant eu aucun effet sur la réponse pharmacologique et n'a entraîné aucun effet indésirable.
Génotoxicité
Aucune étude n'a été réalisée pour établir le pouvoir génotoxique du satralizumab. Les anticorps ne devraient pas avoir d'effets sur l'ADN.
Carcinogénicité
Aucune étude de carcinogénicité n'a été réalisée avec le satralizumab. Aucune lésion proliférative n'a été observée dans une étude de toxicité chronique de 6 mois chez le singe cynomolgus.
Fertilité
Chez le singe, l'administration prolongée de satralizumab n'a eu aucun effet sur les organes de reproduction mâles et femelles.
Toxicité sur la reproduction
Le traitement de guenons cynomolgus gestantes par des doses maximales de 50 mg de satralizumab par kg et par semaine jusqu'à la mise bas n'a entraîné aucun effet délétère sur les mères, le développement fœtal, l'issue de la gestation ou la survie et le développement des jeunes animaux, y compris la capacité d'apprentissage. La réponse anticorps à un antigène thymo-dépendant était plus faible dans la progéniture des animaux traités par le satralizumab que dans la progéniture des animaux témoins, ce qui est vraisemblablement en rapport avec l'effet pharmacologique sur l'IL-6.
La progéniture des mères traitées par le satralizumab a présenté une exposition systémique et des taux plasmatiques élevés d'IL-6 pendant jusqu'à 6 mois après la naissance. Le satralizumab a été détecté dans le lait (<0,9% de la concentration plasmatique respective de la mère).
Remarques particulières
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C).
Non ouvert, Enspryng peut être sorti du réfrigérateur et y être remis si cela est nécessaire. Il ne doit pas être conservé pendant plus de 8 jours hors du réfrigérateur à température ambiante (au max. 30 °C).
Ne pas agiter. Ne pas congeler.
Conserver le récipient dans son carton pour le protéger de la lumière.
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Enspryng est à usage unique.
Avant l'emploi, conserver la seringue préremplie 30 minutes à température ambiante hors de sa boîte.
Ne pas injecter le médicament si le liquide est trouble, a une couleur anormale ou contient des particules.
Vérifier que la seringue préremplie et le protège-aiguille ne sont pas endommagés. Ne pas les utiliser en présence de fissures ou de défectuosités.
Élimination de la seringue préremplie et du protège-aiguille
Suivre impérativement les consignes suivantes concernant l'utilisation et l'élimination de la seringue préremplie et du protège-aiguille:
- Ne réutiliser en aucun cas la seringue préremplie.
- Placer la seringue usagée dans un récipient pour objets pointus et tranchants immédiatement après l'utilisation.
- Éliminer la seringue préremplie et le protège-aiguille conformément à la règlementation locale en vigueur ou aux instructions du personnel médical.
- Conserver la seringue préremplie, le protège-aiguille ainsi que tous les médicaments hors de portée des enfants.
Élimination des médicaments non utilisés/périmés
Le rejet de médicaments dans l'environnement doit être réduit au minimum. Les médicaments ne doivent pas être éliminés avec les eaux usées. Éviter toute élimination avec des déchets ménagers. Utilisez des «systèmes collecteurs» établis s'ils sont disponibles sur votre site.
Numéro d’autorisation
67617 (Swissmedic).
Titulaire de l’autorisation
Roche Pharma (Suisse) SA, Bâle.
Mise à jour de l’information
Juillet 2020.
Відгуки (0)

Безкоштовна консультація досвідченого спеціаліста
Опишіть симптоми або потрібний продукт - ми допоможемо підібрати його дозування або аналог, оформити замовлення з доставкою додому або просто проконсультуємо.
Нас 14 спеціалістів і 0 ботів. Ми завжди будемо з вами на зв'язку і зможемо зв'язатися в будь-який час.
Хіт продажів


HerpoTherm ручка від герпесу
Код продукту: 7798882Herpotherm® - нагрівальна ручка Герпез не тільки непривабливий, але й може бути дуже болючим. Супут..
8214.70 RUB


Burgerstein iron plus капсули Ds 150 шт
Код продукту: 3032006і назвати важливі властивості товару. Burgerstein iron plus капсули Ds 150 шт Капсули Burgerstein ir..
6397.40 RUB


Extra Cell Matrix C-II TABS для суглобів 120 шт
Код продукту: 5677150Extra Cell Matrix C-II TABS для суглобів 120 шт Extra Cell Matrix C-II TABS for Joints – це високоеф..
13106.98 RUB






Vita Omexanthin капсули 60 шт
Код продукту: 6161945Vita Omexanthin капсули - 60 шт Капсули Vita Omexanthin - це дієтична добавка з комбінацією омега-3 ..
16063.71 RUB




Альгіфор-Л форте таблетки плівка 400 мг 10 шт
Код продукту: 3398902..
3515.40 RUB



Зубна щітка Trisa Flexible Head жорстка
Код продукту: 2841175Зубна щітка Trisa Flexible Head жорстка Жорстка зубна щітка Trisa Flexible Head ідеально підходить д..
989.74 RUB





Пастилки Phytopharma Islandica без цукру 40 шт
Код продукту: 2561234Пастилки Phytopharma Islandica без цукру 40 шт Опис товару: Пастилки Phytopharma Islandica без цукру..
1808.99 RUB

