

Ремодулин Инф Лёс 2,5 мг/мл Флакон 20 мл
Remodulin Inf Lös 2.5 mg/ml Vial 20 ml
-
897179.82 RUB
При оплате криптовалютой:
Ваша прибыль 89717.98 RUB / 1048.75 USDT

- Наличие: Нет в наличии
- Производитель: ORPHA SWISS GMBH
- Модель: 2927161
- ATC-код B01AC21
- EAN 7680561200038
Состав:
Наведите телефон на qr-код

Описание
 Deutsch
Deutsch French
French Italian
ItalianWas ist Remodulin und wann wird es angewendet?
Auf Verschreibung des Arztes oder der Ärztin.
Remodulin enthält den Wirkstoff Treprostinil (als Natrium-Treprostinil) und wird zur Behandlung bestimmter Formen der pulmonalen arteriellen Hypertonie eingesetzt (Bluthochdruck in den Arterien zwischen Herz und Lunge).
Wann darf Remodulin nicht angewendet werden?
Remodulin darf bei Allergie gegenüber einem Bestandteil von Remodulin oder ähnlichen Arzneimitteln nicht angewendet werden.
Remodulin darf auch nicht angewendet werden,
- wenn bei Ihnen eine «pulmonale veno-okklusive Erkrankung» diagnostiziert wurde. Dies ist eine Erkrankung, bei der die Blutgefässe, die Blut durch die Lungen transportieren, anschwellen und verstopfen, was zu erhöhtem Blutdruck in den Blutgefässen zwischen Herz und Lunge führt;
- wenn Sie Herzprobleme haben, wie z.B.:
- Herzschwäche,
- diagnostizierter Herzfehler, wie z.B. eine fehlerhafte Herzklappe, die die Funktion des Herzens beeinträchtigt,
- schwere Erkrankung der Herzkranzgefässe oder instabile Angina Pectoris,
- Myokardinfarkt (Herzinfarkt) innerhalb der letzten sechs Monate,
- schwere Herzrhythmusstörungen,
- Herzerkrankungen, die nicht behandelt oder ärztlich kontrolliert werden;
- wenn Sie an einer schweren Lebererkrankung leiden;
- wenn bei Ihnen ein besonders hohes Blutungsrisiko besteht, wie z.B. bei aktiven Magengeschwüren, bei Verletzungen und anderen Blutungen;
- wenn Sie innerhalb der letzten 3 Monate einen Schlaganfall oder andere Durchblutungsstörungen des Gehirns hatten.
Wann ist bei der Anwendung von Remodulin Vorsicht geboten?
Informieren Sie Ihren Arzt bzw. Ihre Ärztin vor Beginn der Behandlung, wenn Sie an einer der folgenden Erkrankungen/Zustände leiden oder eines dieser Arzneimittel einnehmen/anwenden:
- Leber- oder Nierenfunktionsstörungen,
- starkes Übergewicht (Body-Mass-Index (BMI) über 30 kg/m2),
- Infektion mit dem humanen Immundefizienz-Virus (HIV),
- erhöhter Blutdruck in den Lebervenen (portale Hypertonie),
- angeborener Herzfehler, der den Blutfluss in Ihrem Herzen beeinträchtigt,
- bei natriumarmer Diät,
- bei gleichzeitiger Anwendung von Arzneimittel wie Antigerinnungsmittel (Antikoagulanzien oder Thrombozyten-Aggregationshemmer) oder Arzneimittel zur Behandlung von Schmerzen oder rheumatischen Beschwerden, Kortisonpräparate, Mittel zur Behandlung von zu hohem Blutdruck, gefässerweiternde Mittel, harntreibende Arzneimittel, gewisse Arzneimittel, welche die Wirkung von Remodulin vermindern oder verstärken können (z.B. Arzneimittel mit den Wirkstoffen Gemfibrozil, Trimethoprim, Deferasirox, Rifampicin, Phenytoin, Carbamazepin, Phenobarbital oder Johanniskraut).
Beachten Sie strikt die Anweisungen, die Sie zur Herstellung und Verabreichung von Remodulin erhalten haben.
Informieren Sie Ihren Arzt bzw. Ihre Ärztin während der Behandlung mit Remodulin,
- wenn Ihr Blutdruck abfällt (Hypotonie); dies kann sich durch das Auftreten von Schwindel oder Ohnmacht äussern,
- wenn es bei Ihnen über einen kurzen Zeitraum zu deutlich stärkerer Atemnot oder beständigem Husten kommt,
- wenn bei Ihnen Blutungen auftreten (z.B. Nasenbluten, Zahnfleischbluten, Blut im Stuhl),
- wenn es bei Ihnen zu Fieber kommt, während Sie Remodulin intravenös erhalten oder die intravenöse Infusionsstelle sich rötet, anschwillt und/oder schmerz- und druckempfindlich wird, da dies ein Zeichen einer Infektion sein kann.
Dieses Arzneimittel kann die Reaktionsfähigkeit, die Fahrtüchtigkeit und die Fähigkeit, Maschinen zu bedienen, beeinträchtigen! Vor allem bei Behandlungsbeginn oder nach Dosisanpassungen können ein tiefer Blutdruck oder Schwindel auftreten. In diesen Fällen dürfen Sie sich nicht an das Steuer eines Fahrzeugs setzen bzw. Werkzeuge oder Maschinen bedienen.
Remodulin enthält max. 75 mg Natrium (Dosisstärke 5 mg/ml) pro 20 ml Durchstechflasche, entsprechend 3.75% der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g. Das muss bei Patienten mit einer kontrollierten Natriumdiät in Betracht gezogen werden.
Informieren Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, wenn Sie an anderen Krankheiten leiden, Allergien haben oder andere (auch selbstgekaufte!) Arzneimittel einnehmen oder anwenden.
Darf Remodulin während einer Schwangerschaft oder in der Stillzeit angewendet werden?
Remodulin darf nur nach Absprache mit dem Arzt bzw. der Ärztin während der Schwangerschaft angewendet werden. Wenn Sie während der Behandlung mit Remodulin schwanger werden, suchen Sie rasch möglichst Ihren Arzt bzw. Ihre Ärztin auf.
Während der Behandlung mit Remodulin sollte nicht gestillt werden.
Wie verwenden Sie Remodulin?
Die Behandlung mit Remodulin wird im Spital unter ärztlicher Überwachung begonnen. Remodulin wird als subkutane oder intravenöse Dauerinfusion appliziert.
Subkutane Dauerinfusion:
Die Kanüle wird unter Haut auf dem Bauch oder auf dem Oberschenkel platziert und befestigt. Remodulin wird über eine kleine Pumpe, die am Gurt, an der Wade, am Oberschenkel oder Bauch befestigt wird, dauernd mit konstantem Fluss freigesetzt. Die Remodulin Durchstechflasche ist Teil der Pumpe, und die Lösung wird über einen feinen Polyvinyl-Schlauch zur Nadel transportiert.
Intravenöse Dauerinfusion:
Im Spital wird Ihnen ein zentraler Venenkatheder gesetzt. Über diesen wird die verdünnte Remodulin-Lösung mittels einer Pumpe dauernd infundiert. Die Lösung von 50 oder 100 ml wird über maximal 48 Stunden verabreicht werden. Danach wird die Lösung durch eine frische Verdünnung ersetzt.
Anwendung:
Im Spital wird Ihnen vom Personal der Gebrauch der Pumpe, das Einsetzen des Infusionsbestecks, der Ersatz von Remodulin und der Einsatz der Dauerinfusion genau erklärt.
Die Durchflussrate von Remodulin wird jede Woche neu eingestellt.
Meistens wird die Dosierung in Abhängigkeit der Symptome der pulmonalen Hypertonie leicht erhöht. Falls unerträgliche Nebenwirkungen auftreten, wird die Dosierung nach Anweisung Ihres Arztes bzw. Ihrer Ärztin angepasst.
Für die korrekte Handhabung der Infusionspumpe halten Sie sich bitte an die Gebrauchsanweisung der Pumpe.
Vergewissern Sie sich, dass Sie die richtige Remodulin-Konzentration und das richtige Infusionsbesteck anwenden und wechseln Sie alle 48 bis 72 Stunden (bei der subkutanen Dauerinfusion) bzw. mindestens alle 48 Stunden (bei der intravenösen Dauerinfusion) das Infusionsbesteck und das entsprechende Arzneimittelreservoir. Beachten Sie die hygienische Handhabung.
Ein schlechtes Funktionieren der Pumpe oder eine Verstopfung des Infusionsbestecks können zu einer Verschlechterung der Symptome der pulmonalen arteriellen Hypertonie führen. Im Falle eines Unterbruchs des Flusses von Remodulin halten Sie sich an die Anweisungen der Bedienungsanleitung der Pumpe und verständigen Sie – falls nötig – Ihren Arzt bzw. Ihre Ärztin oder die Klinik. Halten Sie immer eine Infusionspumpe, Infusionsbesteck und Remodulin in Reserve.
Die Anwendung und Sicherheit von Remodulin bei Kindern und Jugendlichen ist bisher nicht geprüft worden.
Ihr Arzt bzw. Ihre Ärztin wird die Dosierung und Behandlungsdauer an Ihre persönliche Situation anpassen.
Ändern Sie nicht von sich aus die verschriebene Dosierung. Wenn Sie glauben, das Arzneimittel wirke zu schwach oder zu stark, so sprechen Sie mit Ihrem Arzt oder Apotheker bzw. mit Ihrer Ärztin oder Apothekerin.
Überdosierung:
Im Falle einer Überdosierung könnten folgende Symptome auftreten: Hitzewallungen, Kopfschmerzen, Übelkeit, Erbrechen, Durchfall und eine Senkung des Blutdrucks. Es wird empfohlen, unter Anleitung Ihres Arztes bzw. Ihrer Ärztin die Durchflussrate zu senken oder zu unterbrechen bis die Symptome wieder verschwunden sind. Danach kann wieder mit der vom Arzt bzw. der Ärztin verschriebenen Dosierung weitergefahren werden.
Welche Nebenwirkungen kann Remodulin haben?
Die meisten Nebenwirkungen treten an der Einstichstelle der Kanüle auf (85%). Schmerzen, Röte, Hitze, Schwellung und Blutungen zeigen sich oft zu Beginn der Behandlung, im Verlaufe der Behandlung aber weniger häufig. Ihr Arzt bzw. Ihre Ärztin kann die Dosierung von Remodulin senken, wenn Ihre Nebenwirkungen sehr ernsthaft sind.
Eine Interpretation der übrigen Nebenwirkungen ist dadurch erschwert, dass einzelne klinische Symptome der pulmonalen arteriellen Hypertonie (z.B. Schwindel, Ohnmacht) ähnlich sind. Nebenwirkungen, die wahrscheinlich auf die zugrunde liegende Krankheit zurückzuführen sind, schliessen Atemnot, Müdigkeit, Brustschmerzen und Blässe ein.
Sehr häufig (betrifft mehr als einen von 10 Anwendern)
Schmerzen an der Infusionsstelle (85%), Reaktionen an der Infusionsstelle (83%), Kopfschmerzen (30%), Durchfall (22%), Blutungen (vor allem bei Patienten, die gleichzeitig mit blutverdünnenden Arzneimitteln behandelt werden) oder blaue Flecken an der Infusionsstelle (21%), Übelkeit (19%), Gefässerweiterung / Hitzewallungen (11%), Kieferschmerzen (13%), Hautausschlag (12%).
Häufig (betrifft 1 bis 10 von 100 Anwendern)
Schwindel, tiefer Blutdruck, Erbrechen, Juckreiz, Muskel- und Gelenkschmerzen, Schmerzen in Armen und / oder Beinen, Schwellungen (Ödeme) sowie Blutungsereignisse wie z.B. Nasenbluten, Bluthusten, Blut im Urin, Zahnfleischbluten, Blut im Stuhl auf.
Es wurden auch Infektionen oder Abszesse an der Infusionsstelle, Blutungen an der Infusionsstelle, Entzündungen des Gewebes unter der Haut, eine verminderte Anzahl Blutplättchen, Herzmuskelschwäche bei hohem Volumen des Blutes, das pro Zeitspanne vom Herzen gepumpt wird, mit der Folge von Kurzatmigkeit, Ermüdung, Schwellung der Beine- und des Bauchraums sowie anhaltendem Husten, Hautausschläge, Knochenschmerzen beobachtet.
Im Zusammenhang mit der intravenösen Gabe wurde zusätzlich über Schmerzen an der Eintrittsstelle des Katheters, Venenentzündung, Infektionen des Blutkreislaufes und schwere bakterielle Blutinfektion (Sepsis), Bildung von Blutpfropfen mit Verschluss von Gefässen sowie Störungen des Verabreichungssystems berichtet.
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Dies gilt insbesondere auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind.
Was ist ferner zu beachten?
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Aufbrauchfrist nach Anbruch
Einmal angestochene Durchstechflaschen dürfen nicht länger als 30 Tage verwendet werden.
Die verdünnte Lösung für die intravenöse Infusion darf maximal über 48 Stunden verabreicht werden. Danach sind allfällige Reste zu vernichten und eine neue Verdünnung zu verwenden.
Lagerungshinweis
Das ungeöffnete Arzneimittel bei Raumtemperatur (15- 25 °C) und ausserhalb der Reichweite von Kindern lagern.
Weitere Auskünfte erteilt Ihnen gern Ihr Arzt oder Apotheker bzw. Ärztin oder Apothekerin. Diese Personen verfügen über die ausführliche Fachinformation.
Was ist in Remodulin enthalten?
Wirkstoffe
Treprostinil (als Treprostinil-Natrium) in folgenden Konzentrationen: 1 mg/ml, 2.5 mg/ml, 5 mg/ml und 10 mg/ml.
Hilfsstoffe
Natriumcitrat-Dihydrat, Salzsäure, Metacresol, Natriumhydroxid, Natriumchlorid, Wasser für Injektionszwecke.
Zulassungsnummer
56120 (Swissmedic)
Wo erhalten Sie Remodulin? Welche Packungen sind erhältlich?
In Apotheken nur gegen ärztliche Verschreibung.
Remodulin 1 mg/ml, 2.5 mg/ml, 5 mg/ml, 10 mg/ml: Einzelpackungen mit 1 Durchstechflasche zu 20 ml
Zulassungsinhaberin
Gebro Pharma AG, 4410 Liestal
Diese Packungsbeilage wurde im Juni 2020 letztmals durch die Arzneimittelbehörde (Swissmedic) geprüft.
Qu'est-ce que Remodulin et quand doit-il être utilisé?
Selon prescription du médecin.
Remodulin contient le principe actif tréprostinil (sous forme de tréprostinil sodique) et est utilisé pour le traitement de certaines formes d'hypertension artérielle pulmonaire (élévation de la tension sanguine dans les artères situées entre le cœur et les poumons).
Quand Remodulin ne doit-il pas être pris/utilisé?
Remodulin ne doit pas être utilisé en cas d'allergie à un composant de Remodulin ou à des médicaments similaires.
Remodulin ne doit pas non plus être utilisé
- Si une «maladie veino-occlusive pulmonaire» a été diagnostiquée chez vous. Cette maladie se caractérise par le gonflement et l'occlusion des vaisseaux sanguins transportant le sang à travers les poumons, ce qui entraîne une augmentation de la pression artérielle dans les vaisseaux sanguins situés entre le cœur et les poumons;
- Si vous avez un problème cardiaque tel que:
- Insuffisance cardiaque
- Malformation cardiaque connue, telle que valvulopathie entraînant un dysfonctionnement du cœur
- Maladie grave des artères coronaires ou angor instable
- Infarctus du myocarde survenu au cours des 6 derniers mois
- Anomalies graves du rythme cardiaque
- Toutes maladies du cœur non traitées ou instables
- Si vous souffrez de troubles sévères de la fonction hépatique;
- Si vous présentez d'importants facteurs de risque d'hémorragie tels qu'un ulcère gastro-intestinal actif, des blessures ou tout autre état hémorragique;
- Si vous avez eu un accident vasculaire cérébral au cours des 3 derniers mois, ou toute autre affection ayant conduit à une modification du débit sanguin au niveau cérébral.
Quelles sont les précautions à observer lors de la prise/de l'utilisation de Remodulin?
Informez votre médecin avant le début du traitement si vous souffrez de l'une des maladies ou de l'un des états suivants ou si vous prenez l'un de ces médicaments:
- Insuffisance hépatique ou rénale
- Surcharge pondérale (indice de masse corporelle [IMC] supérieur à 30 kg/m2)
- Infection au virus de l'immunodéficience humaine (VIH)
- Pression artérielle élevée au niveau des veines du foie (hypertension portale)
- Cardiopathie congénitale qui perturbe la circulation sanguine à travers le cœur
- Régime hyposodé
- Prise simultanée de médicaments tels que des fluidifiants sanguins (anticoagulants ou inhibiteurs de l'agrégation plaquettaire) ou des médicaments pour le traitement de la douleur ou des troubles rhumatismaux, des médicaments à base de cortisone, des médicaments utilisés pour traiter l'hypertension artérielle, des vasodilatateurs, des diurétiques, certains médicaments susceptibles d'augmenter ou de diminuer l'effet de Remodulin (exemple: médicaments contenant les principes actifs gemfibrozil, triméthoprime, déférasirox, rifampicine, phénytoïne, carbamazépine, phénobarbital ou millepertuis)
Respectez scrupuleusement les consignes que vous avez reçues pour la préparation et l'administration de Remodulin.
Au cours du traitement par Remodulin, informez votre médecin
- si votre pression artérielle diminue (hypotension); cela peut se manifester par des vertiges ou une perte de connaissance;
- si vous éprouvez une aggravation rapide de vos difficultés respiratoires ou une toux persistante;
- si vous présentez des saignements (exemple: saignement du nez ou des gencives, sang dans les selles);
- si vous présentez de la fièvre pendant l'administration intraveineuse de Remodulin ou si la zone d'insertion de la perfusion intraveineuse devient rouge, gonflée et/ou douloureuse au toucher, car cela pourrait être le signe d'une infection.
Ce médicament peut affecter les réactions, l'aptitude à conduire et la capacité à utiliser des machines! L'initiation du traitement ou les phases d'ajustement de la posologie peuvent s'accompagner d'une baisse de pression artérielle ou de vertiges. Dans ce cas, vous ne devez pas conduire de véhicules ni utiliser d'outils ou de machines.
Remodulin contient au max. 75 mg (dosage de 5 mg/ml) de sodium par flacon de 20 ml, ce qui équivaut à 3,75% de l'apport alimentaire quotidien maximal recommandé par l'OMS de 2 g de sodium par adulte. Cela doit être pris en considération chez les patients suivant un régime hyposodé strict.
Veuillez informer votre médecin ou votre pharmacien si vous souffrez d'une autre maladie ou d'allergie, ou si vous prenez déjà d'autres médicaments en usage interne ou externe (même en automédication!).
Remodulin peut-il être pris/utilisé pendant la grossesse ou l'allaitement?
Remodulin ne peut être utilisé pendant la grossesse qu'après avoir demandé l'avis du médecin. Si vous tombez enceinte pendant le traitement par Remodulin, consultez le plus rapidement possible votre médecin.
Il ne faut pas allaiter pendant le traitement par Remodulin.
Comment utiliser Remodulin?
Le traitement par Remodulin est commencé à l'hôpital sous surveillance médicale. Remodulin est administré en perfusion sous-cutanée ou intraveineuse continue.
Perfusion sous-cutanée continue:
La canule est placée et fixée sous la peau, sur le ventre ou sur la cuisse. Remodulin est libérée en continu avec un débit constant, par une petite pompe qui est fixée à la ceinture, au mollet, à la cuisse ou au ventre. Le flacon en verre de Remodulin est un élément de la pompe et la solution est transportée vers l'aiguille par un fin tuyau en polyvinyle.
Perfusion intraveineuse continue:
À l'hôpital on vous mettra en place un cathéter veineux par lequel se fera, au moyen d'une pompe, la perfusion continue de la solution diluée de Remodulin. La solution de 50 ou 100 ml sera administrée en l'espace de 48 heures au maximum. Elle sera alors remplacée par une dilution nouvellement préparée.
Utilisation:
À l'hôpital, le personnel vous expliquera précisément comment utiliser la pompe, mettre en place le set de perfusion, remplacer Remodulin et employer la perfusion continue. Le débit de Remodulin sera fixé de nouveau chaque semaine.
Le plus souvent, le dosage sera légèrement augmenté en fonction des symptômes de l'hypertension pulmonaire. Si des effets secondaires intolérables surviennent, le dosage sera ajusté selon les instructions de votre médecin.
Veuillez respecter le mode d'emploi de la pompe de perfusion pour assurer le maniement correct de celle-ci.
Assurez-vous que vous utilisez la concentration correcte de Remodulin et le bon set de perfusion et changez le set de perfusion et le réservoir correspondant de médicament toutes les 48 à 72 heures (en cas de perfusion sous-cutanée continue) ou au minimum toutes les 48 heures (en cas de perfusion intraveineuse continue). Veuillez respecter les règles d'hygiène lors du maniement.
Un mauvais fonctionnement de la pompe ou une obstruction du set de perfusion peuvent entraîner une aggravation des symptômes de l'hypertension artérielle pulmonaire. En cas d'interruption de l'écoulement de Remodulin, observez les instructions du mode d'emploi de la pompe et avertissez – si nécessaire – votre médecin ou la clinique. Gardez toujours une pompe de perfusion, un set de perfusion et un flacon de Remodulin en réserve.
L'utilisation et la sécurité d'emploi de Remodulin chez l'enfant et l'adolescent n'a pas fait l'objet d'études à ce jour.
Votre médecin ajustera le dosage et la durée du traitement à votre situation personnelle.
Ne changez pas de votre propre chef le dosage prescrit.
Adressez-vous à votre médecin ou à votre pharmacien si vous estimez que l'efficacité du médicament est trop faible ou au contraire trop forte.
Surdosage
En cas de surdosage, les symptômes suivants peuvent apparaître: bouffées vasomotrices, maux de tête, nausées, vomissements, diarrhée et baisse de la tension artérielle. Il est recommandé, sous la direction de votre médecin, de diminuer le débit ou d'arrêter la perfusion jusqu'à ce que les symptômes aient disparu. La perfusion peut ensuite être poursuivie avec le dosage prescrit par le médecin.
Quels effets secondaires Remodulin peut-il provoquer?
La plupart des effets indésirables surviennent à l'endroit de la ponction de la canule (85%). Des douleurs, une rougeur, une chaleur, un gonflement et des saignements se produisent souvent au début du traitement, mais deviennent moins fréquents au cours du traitement. Votre médecin pourra diminuer le dosage de Remodulin si vous souffrez d'effets secondaires très sévères.
L'interprétation des autres effets secondaires est compliquée par le fait que certains symptômes cliniques de l'hypertension artérielle pulmonaire (par ex. vertiges, évanouissement) sont similaires. Les effets indésirables pouvant vraisemblablement être rattachés à la maladie sous-jacente comprennent difficultés respiratoires, fatigue, douleurs dans la poitrine et pâleur.
Très fréquent (concerne plus d'un utilisateur sur 10)
Douleurs au site de perfusion (85%), réactions au site de perfusion (83%), maux de tête (30%), diarrhée (22%), saignements (principalement chez les patients sous traitement simultané par anticoagulants) ou bleus au niveau du site de perfusion (21%), nausées (19%), dilatation de vaisseaux / bouffées vasomotrices (11%), douleurs dans la mâchoire (13%), éruptions cutanées (12%).
Fréquent (concerne 1 à 10 utilisateurs sur 100)
Vertiges, une baisse de pression artérielle, les vomissements, les démangeaisons, les douleurs musculaires et articulaires, douleurs aux jambes et/ou bras, les gonflements (œdèmes) ainsi que les incidents hémorragiques tels que les saignements du nez, l'hémoptysie, le sang dans l'urine, le saignement gingival ou le sang dans les selles.
Des infections ou abcès au site de perfusion, des saignements au site de perfusion, une inflammation du tissu situé sous la peau, une diminution du nombre de plaquettes, l'augmentation du travail du cœur pour pomper le sang peut entraîner essoufflement, fatigue, œdèmes des jambes et de l'abdomen en raison d'une accumulation de liquide, toux persistante, des éruptions cutanées et des douleurs osseuses ont également été observés.
Lors de l'administration par voie intraveineuse, des douleurs au site d'insertion du cathéter, une inflammation veineuse, des infections touchant la circulation sanguine et une infection bactérienne généralisée grave (septicémie), la formation de caillots sanguins avec occlusion vasculaire ainsi que des dysfonctionnements du système d'administration ont été rapportés.
Si vous remarquez des effets secondaires, veuillez en informer votre médecin ou votre pharmacien. Ceci vaut en particulier pour les effets secondaires non mentionnés dans cette notice d'emballage.
À quoi faut-il encore faire attention?
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention <EXP> sur le récipient.
Délai d'utilisation après ouverture
Les flacons une fois percés peuvent s'utiliser pendant 30 jours au maximum.
La solution diluée pour la perfusion intraveineuse peut être administrée pendant 48 heures au maximum. Tout reste doit ensuite être jeté et une nouvelle dilution doit être utilisée.
Remarques concernant le stockage
Conserver le médicament non ouvert à température ambiante (15-25°C) et hors de portée des enfants.
Pour de plus amples renseignements, consultez votre médecin ou votre pharmacien, qui disposent d'une information détaillée destinée aux professionnels.
Que contient Remodulin?
Principes actifs
Tréprostinil (sous forme de tréprostinil sodique) dans les concentrations suivantes: 1 mg/ml, 2.5 mg/ml, 5 mg/ml et 10 mg/ml.
Excipients
Citrate de sodium dihydraté, acide chlorhydrique, métacrésol, hydroxyde de sodium, chlorure de sodium, eau pour préparations injectables.
Numéro d'autorisation
56120 (Swissmedic)
Où obtenez-vous Remodulin? Quels sont les emballages à disposition sur le marché?
En pharmacie, sur ordonnance médicale.
Remodulin 1 mg/ml, 2.5 mg/ml, 5 mg/ml et 10 mg/ml: emballages uniques contenant 1 flacon en verre (vial) de 20 ml.
Titulaire de l'autorisation
Gebro Pharma AG, CH-4410 Liestal
Cette notice d'emballage a été vérifiée pour la dernière fois en juin 2020 par l'autorité de contrôle des médicaments (Swissmedic).
Che cos'è il Remodulin e quando si usa?
Su prescrizione medica.
Remodulin contiene il principio attivo treprostinil (sotto forma di treprostinil-sodio) e viene usato per il trattamento di determinate forme di ipertensione arteriosa polmonare (elevata pressione del sangue nelle arterie tra cuore e polmone).
Quando non si può assumere/usare Remodulin?
Remodulin non deve essere usato in caso di allergia verso un componente di Remodulin o altro medicamento simile.
Allo stesso modo non si può usare Remodulin nei casi seguenti:
- se le è stata diagnosticata una «malattia veno-occlusiva polmonare». Questa malattia interessa i piccoli vasi polmonari che trasportano il sangue attraverso i polmoni, facendoli gonfiare e occludere, e causa un aumento di pressione nei vasi tra cuore e polmone;
- se ha problemi cardiaci come ad es.:
- insufficienza cardiaca,
- difetto cardiaco diagnosticato, ad es. un difetto valvolare che compromette la funzione del cuore,
- grave malattia dei vasi coronarici o angina pectoris instabile,
- infarto miocardico (infarto cardiaco) nei sei mesi precedenti,
- gravi disturbi del ritmo cardiaco,
- malattie cardiache non soggette a trattamento o controllo medico;
- se soffre di una grave malattia del fegato;
- se ha un rischio particolarmente elevato di emorragie, ad es. in presenza di ulcera gastrica attiva, lesioni e altri sanguinamenti;
- se nei 3 mesi precedenti ha avuto un ictus cerebrale o altri disturbi di circolazione al cervello.
Quando è richiesta prudenza nella somministrazione/nell'uso di Remodulin?
Informi il suo medico prima di iniziare il trattamento se soffre di malattie/condizioni tra le seguenti oppure se assume/impiega uno dei medicinali qui indicati:
- alterazioni della funzione epatica o renale,
- forte sovrappeso (Body-Mass-Index (BMI) superiore a 30 kg/m2),
- infezione da virus dell'immunodeficienza umana (HIV),
- pressione sanguigna elevata nelle vene epatiche (ipertensione portale),
- difetto cardiaco congenito che compromette il flusso ematico nel cuore,
- dieta povera di sodio,
- uso concomitante di farmaci inibenti la coagulazione (anticoagulanti oppure antiaggreganti delle piastrine), medicinali antidolorifici o per il trattamento di disturbi reumatici, preparati a base di cortisone, sostanze per il trattamento dell'ipertensione arteriosa, farmaci vasodilatatori, diuretici, determinati medicamenti che possono diminuire o rafforzare l'efficacia di Remodulin (ad es. medicamenti con principio attivo gemfibrozil, trimetoprim, deferasirox, rifampicina, fenitoina, carbamazepina, fenobarbital o iperico.
Segua attentamente le istruzioni ricevute per la preparazione e la somministrazione di Remodulin.
Durante il trattamento con Remodulin informi il suo medico se:
- ha abbassamenti di pressione (ipotensione). L'abbassamento di pressione può manifestarsi con vertigini o svenimento,
- se in un breve arco di tempo sopraggiunge forte difficoltà respiratoria o tosse persistente,
- se sopraggiungono sanguinamenti (ad es. dal naso, sanguinamento gengivale, sangue nelle feci),
- se insorge febbre durante la terapia per via endovenosa con Remodulin oppure se il sito di infusione per la somministrazione endovenosa si arrossa, gonfia e/o diviene dolente e sensibile al tatto, perché può trattarsi di un'infezione.
Questo medicamento può ridurre la capacità di reazione, la capacità di condurre un veicolo e la capacità di utilizzare attrezzi o macchine! Soprattutto all'inizio del trattamento o dopo un adeguamento del dosaggio possono insorgere abbassamento della pressione o vertigini. In questi casi non deve mettersi alla guida di un veicolo e deve evitare di usare attrezzi o macchine.
Remodulin contiene al massimo 75 mg di sodio (dosaggio 5 mg/ml) per flaconcino da 20 ml. Questa quantità equivale al 3,75% dell'assunzione massima giornaliera raccomandata dall'OMS che corrisponde a 2 g di sodio per un adulto. Occorre tenere presente questo dato nei pazienti che seguono una dieta a contenuto controllato di sodio.
Informi il suo medico o il suo farmacista nel caso in cui soffre di altre malattie, soffre di allergie o assume altri medicamenti (anche se acquistati di sua iniziativa!)
Si può assumere/usare Remodulin durante la gravidanza o l'allattamento?
Remodulin deve essere usato durante la gravidanza soltanto dopo accordi con il medico. Se durante il trattamento con Remodulin dovesse verificarsi una gravidanza, consulti quanto prima il suo medico.
Durante il trattamento con Remodulin deve essere evitato l'allattamento.
Come usare Remodulin?
Il trattamento con Remodulin viene iniziato in ospedale sotto osservazione medica. Remodulin è impiegato sotto forma di infusione sottocutanea o endovenosa continua.
Infusione sottocutanea continua
La cannula viene posizionata e fissata sotto la pelle dell'addome o della coscia. Remodulin viene rilasciato in maniera continuativa, tramite un flusso costante, da una piccola pompa fissata alla cintura, alla caviglia, alla coscia o all'addome. Il flaconcino di vetro di Remodulin è parte integrante della pompa, e la soluzione viene trasportata all'ago attraverso un tubo sottile di polivinile.
Infusione endovenosa continua
In ospedale le sarà posizionato un catetere venoso centrale, attraverso il quale, con l'ausilio di una pompa, le verrà praticata l'infusione continua della soluzione diluita di Remodulin. La soluzione, 50 o 100 ml, le sarà somministrata al massimo in 48 ore, dopodiché la soluzione sarà sostituita da una nuova soluzione diluita.
Applicazione:
Il personale ospedaliero le illustrerà esattamente l'uso della pompa, l'applicazione del set d'infusione, la sostituzione del Remodulin e l'applicazione dell'infusione continua.
Ogni settimana viene nuovamente regolata la portata del flusso di Remodulin.
In genere, il dosaggio viene lievemente incrementato in base ai sintomi dell'ipertensione polmonare. Se dovessero comparire effetti collaterali importanti, il dosaggio verrà adeguato su indicazione del suo medico.
Per quanto riguarda la corretta manipolazione della pompa d'infusione, si prega di attenersi alle istruzioni d'uso della pompa.
Si accerti della correttezza della concentrazione di Remodulin e del set d'infusione che sta utilizzando. Ogni 48-72 ore il set d'infusione e il corrispondente deposito di farmaco vanno sostituiti (se si tratta di infusione continua subcutanea) o almeno ogni 48 ore (se si tratta di infusione continua). Osservi le regole d'igiene durante le manovre.
Un funzionamento deficitario della pompa oppure un'ostruzione del set d'infusione possono determinare un aggravamento dei sintomi dell'ipertensione arteriosa polmonare. Nel caso di un'interruzione del flusso di Remodulin si attenga alle indicazioni fornite nelle istruzioni d'uso della pompa ed avvisi – se necessario – il suo medico oppure la clinica. Tenga sempre di riserva una pompa d'infusione, un set d'infusione e del Remodulin.
Non sono mai state eseguite indagini scientifiche sistematiche sull'applicazione e la sicurezza di Remodulin nei bambini e negli adolescenti finora.
Il suo medico adatterà il dosaggio e la durata del trattamento alla sua personale situazione.
Non modifichi di propria iniziativa la posologia prescritta.
Se ritiene che l'azione del medicamento sia troppo debole o troppo forte ne parli al suo medico o al suo farmacista.
Sovradosaggio
Nel caso di un sovradosaggio potrebbero comparire i seguenti sintomi: vampate di calore, cefalea, nausea, vomito, diarrea e ipotensione arteriosa. Si consiglia di ridurre la portata del flusso secondo l'indicazione del suo medico oppure di interrompere la somministrazione fino alla scomparsa dei sintomi. Successivamente, si può proseguire con il dosaggio prescritto dal medico.
Quali effetti collaterali può avere Remodulin?
Gli effetti collaterali più frequenti si manifestano in corrispondenza della sede di introduzione della cannula (85%). Spesso, all'inizio del trattamento compaiono dolori, arrossamento, calore, tumefazione e sanguinamento che però nel corso del trattamento sono meno frequenti. Se gli effetti collaterali nel suo caso fossero molto seri, il suo medico può ridurre il dosaggio di Remodulin.
Una interpretazione degli ulteriori effetti collaterali è resa difficile dalla loro somiglianza con alcuni sintomi clinici della ipertensione arteriosa polmonare (per es. vertigini, lipotimia). Gli effetti collaterali probabilmente riconducibili alla malattia di base comprendono difficoltà respiratoria, stanchezza, dolori toracici e pallore.
Molto comune (riguarda più di 1 utilizzatore su 10)
Dolori nel sito d'infusione (85%), reazioni nel sito d'infusione (83%), cefalea (30%), diarrea (22%), sanguinamenti (soprattutto nei pazienti contemporaneamente trattati con anticoagulanti) o ecchimosi nella sede d'infusione (21%), nausea (19%) vasodilatazione /vampate di calore (11%), dolori mandibolari (13%) ed eruzione cutanea (12%).
Comune (riguarda da 1 a 10 utilizzatori su 100)
Vertigini, bassa pressione, vomito, prurito, dolori muscolari e articolari, dolore alle gambe e/o alle braccia, gonfiori (edemi) ed episodi di sanguinamento quali sangue dal naso, emissione di sangue con la tosse, presenza di sangue nelle urine, sanguinamento gengivale, sangue nelle feci.
Sono stati anche osservati infezioni o ascessi nel sito d'infusione, sanguinamenti nel sito d'infusione, infezioni del tessuto sottocutaneo, diminuzione delle piastrine, a causa del cuore che pompa troppo sangue si verifica mancanza di respiro, affaticamento, gonfiore delle gambe e dell'addome a causa di un accumulo di liquidi, tosse persistente, eruzioni cutanee e dolori ossei.
Ulteriori effetti collaterali associati alla somministrazione endovenosa segnalati sono: dolori nel punto di inserimento del catetere, infiammazione della vena, infezioni del flusso ematico e grave infezione batterica del sangue (setticemia), formazione di gocce di sangue con occlusione vascolare e malfunzionamenti del sistema di somministrazione.
Se osserva effetti collaterali, si rivolga al suo medico o al suo farmacista, soprattutto se si tratta di effetti collaterali non descritti in questo foglietto illustrativo.
Di che altro occorre tener conto?
Il medicamento non deve essere utilizzato oltre la data indicata con «EXP» sul contenitore.
Termine di consumo dopo l'apertura
Una volta aperti, i flaconcini non devono essere utilizzati per un periodo superiore ai 30 giorni. La soluzione diluita per infusione endovenosa non deve essere somministrata oltre le 48 ore. Successivamente gli eventuali residui devono essere smaltiti e occorre usare una nuova soluzione diluita.
Istruzioni di conservazione
Conservare il medicamento nella confezione integra a temperatura ambiente (15-25 °C) e fuori dalla portata dei bambini.
Il medico, il farmacista che sono in possesso di un'informazione professionale dettagliata, possono darle ulteriori informazioni.
Cosa contiene Remodulin?
Principi attivi
Treprostinil (sotto forma di treprostinil-sodio) nelle seguenti concentrazioni: 1 mg/ml, 2.5 mg/ml, 5 mg/ml e 10 mg/ml.
Sostanze ausiliarie
Sodio citrato diidrato, acido cloridrico, metacresolo, sodio idrossido, sodio cloruro, acqua per preparazioni iniettabili.
Numero dell'omologazione
56120 (Swissmedic)
Dove è ottenibile Remodulin? Quali confezioni sono disponibili?
In farmacia, dietro presentazione della prescrizione medica.
Remodulin 1 mg/ml, 2.5 mg/ml, 5 mg/ml e 10 mg/ml: confezioni singole con 1 flaconcino di vetro (fiala) di 20 ml.
Titolare dell'omologazione
Gebro Pharma AG, CH-4410 Liestal
Questo foglietto illustrativo è stato controllato l'ultima volta nel giugno 2020 dall'autorità competente in materia di medicamenti (Swissmedic).
Zusammensetzung
Wirkstoffe
Treprostinil (als Treprostinil-Natrium).
Hilfsstoffe
Natriumcitrat-Dihydrat, Salzsäure, Metacresol, Natriumhydroxid, Natriumchlorid, Wasser für Injektionszwecke.
Gesamtnatriumgehalt pro mL
Dosierungsstärke | 1.0 | 2.5 | 5.0 | 10.0 |
Gesamtnatriumgehalt (in mg) | 3.7 | 3.75 | 3.92 | 3.74 |
Darreichungsform und Wirkstoffmenge pro Einheit
Infusionslösung zur subkutanen oder intravenösen Verabreichung:
Durchstechflaschen mit 1 mg Treprostinil/ml; 2.5 mg Treprostinil/ml; 5 mg Treprostinil/ml; 10 mg Treprostinil/ml
Indikationen/Anwendungsmöglichkeiten
Langzeitbehandlung von primärer pulmonaler Hypertonie und pulmonal arterieller Hypertonie mit Bindegewebserkrankung bei Patienten mit NYHA III und IV (Einteilung nach der New York Heart Association).
Dosierung/Anwendung
Die Behandlung mit Remodulin wird im Spital unter ärztlicher Überwachung begonnen. Remodulin wird als subkutane oder intravenöse Dauerinfusion appliziert. Aufgrund der Risiken im Zusammenhang mit zentralen Dauervenenverweilkathetern, einschliesslich schwerer Bakteriämien und Sepsis, die tödlich verlaufen können, ist die subkutane Infusion (unverdünnt) die bevorzugte Art der Verabreichung.
Therapieeinleitung
Initialdosis
Die Infusionsrate sollte zu Beginn bei 1.25 ng/kg/min liegen. Falls diese Dosierung vom Patienten nicht vertragen wird, kann die Infusionsrate auf 0.625 ng/kg/min gesenkt werden.
CAVE: Die intravenöse Applikation darf nie unverdünnt erfolgen.
Dosisanpassung/Titration
Das Ziel der chronischen Dosisanpassung ist, die Dosis zu finden, welche die Symptome der pulmonalen arteriellen Hypertonie verbessert, während das Nebenwirkungsprofil erträglich bleibt. In den ersten 4 Wochen sollte die Infusionsrate um 1.25 ng/kg/min pro Woche gesteigert werden und dann um 2.5 ng/kg/min pro Woche.
Dosis-abhängige unerwünschte Wirkungen wie Hitzewallungen (Flushing), Kopfschmerzen, Hypotonie, Übelkeit, Erbrechen und Diarrhöe können eine Reduktion der Infusionsrate notwendig machen, wobei die unerwünschten Wirkungen möglicherweise auch ohne Dosisanpassung wieder verschwinden. Sollte eine unerwünschte Wirkung schlimmer und/oder unerträglich werden, sollte die Infusionsrate gesenkt werden.
Ein plötzlicher Therapieabbruch oder eine starke Reduktion der Dosierung ist zu vermeiden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
In den meisten Fällen kann nach einer Unterbrechung von wenigen Stunden die Infusion in derselben Dosisrate wieder begonnen werden. Bei einer längeren Unterbrechung ist möglicherweise eine Retitrierung der Dosis erforderlich.
In Langzeitanwendungsstudien betrugen die nach 12 Monaten erreichten mittleren Dosisraten 26 ng/kg/min, nach 24 Monaten 36 ng/kg/min und nach 36 Monaten 42 ng/kg/min.
Übliche Dosierung
Subkutane Dauerinfusion
Die subkutane Infusionsrate wird nach folgender Formel berechnet:
Infusionsrate (ml/h):

* Konversionsfaktor (0.00006): 60 min/h × 0.000001 mg/ng
Intravenöse Dauerinfusion
Verdünntes Remodulin wird mittels intravenöser Dauerinfusion über einen Zentralvenenkatheter mittels einer Pumpe für intravenöse Applikation infundiert.
Es kann auch vorübergehend über eine periphere Venenkanüle verabreicht werden, vorzugsweise in eine grosse Vene. Die Anwendung einer peripheren Infusion über mehr als einige Stunden kann das Risiko für Thrombophlebitis erhöhen (siehe «Unerwünschte Wirkungen»).
Die Verwendung eines 0.2 Mikrometer in-line Filters wird empfohlen, um das Risiko einer systemischen Infektion als Folge einer Verunreinigung in der Zubereitung des Medikamentes zu minimieren.
Berechnung der Konzentration, die die verdünnte Remodulin-Lösung haben muss:
Endkonzentration der verdünnten Remodulin-Lösung (mg/ml):

Berechnung der notwendigen Menge Remodulin, die mit ausreichend Verdünnungsmittel (steriles Wasser für Injektionszwecke oder 0.9% NaCl-Lösung für Infusionen) verdünnt werden muss:
Volumen von zu verwendendem Remodulin (ml):

Die berechnete Menge Remodulin wird in das Reservoir gegeben und mit genügend sterilem Wasser für Injektionszwecke oder 0.9% NaCl-Lösung verdünnt.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Die Plasma-Treprostinil-Exposition (Fläche unter der Plasmakonzentrationszeitkurve; AUC) war bei milder bis moderater Leberfunktionsstörung, Child-Pugh-Klasse A bzw. B, um 260% bis 510% erhöht. Milde bis moderate Leberfunktionsstörungen reduzieren die Plasma Clearance von Treprostinil um bis zu 80% verglichen mit gesunden Probanden. Deshalb sollte bei Patienten mit Leberfunktionsstörungen Remodulin nur mit Vorsicht angewendet und der Patient engmaschig bezüglich Nebenwirkungen aufgrund erhöhtem Remodulin überwacht werden (vgl. Pharmakokinetik).
Die initiale Remodulin-Dosis sollte auf 0.625 ng/kg/min reduziert werden und die Dosiserhöhung sollte vorsichtig vorgenommen werden.
In Patienten mit schwerer Leberstörung liegen keine Erfahrungen vor.
Patienten mit Nierenfunktionsstörungen
Es wurden keine Studien mit Patienten mit Nierenfunktionsstörungen durchgeführt. Da Treprostinil und seine Metaboliten primär über die Harnwege ausgeschieden werden, sollten Patienten mit Nierenfunktionsstörungen vorsichtig behandelt werden. Bei diesen Patienten sollte die Dosiserhöhung noch vorsichtiger vorgenommen werden (vgl. Pharmakokinetik).
Anwendung bei Patienten mit Übergewicht
Patienten mit Übergewicht (BMI >30 kg/m²) zeigen eine geringere Clearance von Treprostinil.
Ältere Patienten
Es liegen keine ausreichenden Daten für ältere Patienten (≥65 Jahre) vor. Bei älteren Patienten sollte die Dosierung aufgrund der erhöhten Häufigkeit einer eingeschränkten hepatischen, renalen oder kardialen Funktion sowie von Begleiterkrankungen oder –medikation mit Vorsicht gewählt werden.
Kinder und Jugendliche
Es liegen zu wenige Daten über Kinder und Jugendliche unter 16 Jahren vor, um zu entscheiden, ob die Erwachsenen-Dosierung bei dieser Patientengruppe angewendet werden kann.
Art der Anwendung
Remodulin wird mittels subkutaner oder intravenöser Dauerinfusion verabreicht. Aufgrund der Risiken im Zusammenhang mit zentralen Dauervenenverweilkathetern, einschliesslich schwerer Bakteriämien und Sepsis, die tödlich verlaufen können, ist die subkutane Infusion (unverdünnt) die bevorzugte Art der Verabreichung.
Bei der subkutanen Infusion wird die Kanüle unter die Haut auf dem Bauch oder auf dem Oberschenkel platziert und befestigt. Die Remodulin Durchstechflasche ist bei der subkutanen Applikation Teil der Pumpe. Das Infusionsbesteck und das entsprechende Arzneimittelreservoir muss alle 48 bis 72 Stunden gewechselt werden.
Bei der intravenösen Applikation wird ein zentraler Dauerkatheder gesetzt. Remodulin wird über eine kleine Pumpe, die am Gurt, an der Wade, am Oberschenkel oder Bauch befestigt wird, dauernd mit konstantem Fluss freigesetzt. Für die intravenöse Applikation wird Remodulin verdünnt eingesetzt. Die Lösung wird über einen feinen Polyvinyl-Schlauch zur Nadel transportiert.
Bei der intravenösen Infusion muss die verdünnte Lösung (wegen der Stabilität von Remodulin) zumindest alle 48 Stunden gewechselt werden.
Im Spital wird den Patienten bzw. Patientinnen vom Personal der Gebrauch der Pumpe, das Einsetzen des Infusionsbestecks, der Ersatz des Remodulin-Reservoirs und der Einsatz der Dauerinfusion genau erklärt. Die hygienische Handhabung ist zu beachten.
Die Durchflussrate von Remodulin wird jede Woche neu eingestellt. Meistens wird die Dosierung in Abhängigkeit der Symptome der pulmonalen Hypertonie leicht erhöht. Falls unerträgliche Nebenwirkungen auftreten, wird die Dosierung nach Anweisung des Arztes bzw. der Ärztin angepasst.
Die korrekte Handhabung der Infusionspumpe ist in der Gebrauchsanweisung der Pumpe beschrieben.
Es ist wichtig, dass die richtige Remodulin-Konzentration und das richtige Infusionsbesteck angewendet werden.
Ein schlechtes Funktionieren der Pumpe oder eine Verstopfung des Infusionsbestecks können zu einer Verschlechterung der Symptome der pulmonalen arteriellen Hypertonie führen. Im Falle eines Unterbruchs des Flusses von Remodulin sind die Anweisungen der Bedienungsanleitung der Pumpe einzuhalten. Der Patient bzw. die Patientin sollte immer eine Infusionspumpe, Infusionsbesteck und Remodulin in Reserve halten.
Die Infusionspumpe, mit der Remodulin appliziert wird, sollte folgende Anforderungen erfüllen:
- klein und leicht sein,
- die Anpassung der Infusionsrate in Schritten von 0.002 ml/h für die s.c Infusion sowie von ca. 0.05 ml/Std. für die i.v. Infusion erlauben. Eine typische Infusionsrate für i.v. Infusionspumpen läge zwischen 0.4 und 2 ml pro Stunde.
- sollte Alarmfunktionen bei Verstopfung/fehlendem Fluss, schwacher Batterie, Programmfehler oder Motor-Störungen haben,
- eine Fluss-Genauigkeit von ±6% oder besser aufweisen,
- mit positivem Druck betrieben werden,
- das Reservoir sollte aus Polyvinylchlorid, Polypropylen oder Glas sein.
Remodulin ist in Konzentrationen von 1 mg/ml, 2,5 mg/ml, 5 mg/ml und 10 mg/ml erhältlich.
Die Tabellen als Dosierungshilfen sind am Ende der Fachinformation aufgeführt.
Kontraindikationen
- Überempfindlichkeit gegenüber einem Inhaltsstoff von Remodulin oder gegenüber strukturverwandten Stoffen von Treprostinil,
- pulmonale arterielle Hypertonie in Verbindung mit einer Venenverschlusserkrankung,
- kongestive Herzinsuffizienz infolge einer schweren Funktionsstörung der linken Herzkammer,
- schwere Leberfunktionsstörungen (Child-Pugh-Klasse C),
- aktives Magen-Darm-Geschwür, intrakranielle Blutung, Verletzung oder andere Blutungen,
- kongenitale oder erworbene Herzklappenfehler mit klinisch relevanten Störungen der Myokardfunktion, die nicht mit pulmonaler Hypertonie zusammenhängen,
- schwere koronare Herzkrankheit oder instabile Angina; Herzinfarkt innerhalb der letzten sechs Monate; dekompensierte Herzinsuffizienz, wenn diese nicht unter engmaschiger ärztlicher Überwachung steht; schwere Arrhythmien; zerebrovaskuläre Ereignisse (z.B. transitorische ischämische Attacke, Schlaganfall) innerhalb der letzten drei Monate.
Warnhinweise und Vorsichtsmassnahmen
Allgemeine Vorsichtsmassnahmen
Remodulin sollte nur von Spezialärzten mit Erfahrung in der Diagnose und Behandlung von pulmonaler arterieller Hypertonie eingesetzt werden. Die Entscheidung zum Therapiebeginn mit Remodulin sollte im Wissen erfolgen, dass die Therapie mit grosser Wahrscheinlichkeit über lange Zeit, möglicherweise über Jahre durchgeführt werden muss. Die Fähigkeit des Patienten, Remodulin zu applizieren und das Infusionssystem zu pflegen, sollte berücksichtigt werden.
Remodulin sollte nur subkutan oder in verdünnter Form intravenös verabreicht werden.
Remodulin ist ein stark wirksamer pulmonaler und systemischer Vasodilatator. Die Behandlung mit Remodulin muss daher mit entsprechend ausgebildetem Personal und einer Ausrüstung für eine physiologische Überwachung und Notfallbehandlung begonnen werden. Die Dosisanpassungen wurden in den klinischen Studien aufgrund der Symptome der pulmonalen arteriellen Hypertonie und der Nebenwirkungen von Remodulin vorgenommen.
Risiken aufgrund der intravenösen Verabreichung von Remodulin
Bei Patienten, die Remodulin mittels intravenöser Infusion erhalten haben, wurde über Zentralvenenkatheter-assoziierte Bakteriämien und Sepsis berichtet, die tödlich verlaufen können. Diese Risiken hängen mit dem Verabreichungsweg zusammen. Die subkutane Dauerinfusion (unverdünnt) ist deshalb die bevorzugte Art der Verabreichung. Die intravenöse Dauerinfusion sollte Patienten vorbehalten bleiben, die mit einer subkutanen Treprostinil-Infusion stabilisiert wurden und die die subkutane Gabe nicht vertragen und bei denen diese Gefahren als akzeptabel angesehen werden.
Bei der Herstellung und Verabreichung von Remodulin sind aseptische Techniken einzusetzen.
Risiko einer systemischen Hypotonie
Bei Patienten mit niedrigem systemischem Arteriendruck kann eine Treprostinil-Behandlung die Gefahr einer systemischen Hypotonie erhöhen. Die Behandlung von Patienten mit einem systolischen Arteriendruck von weniger als 85 mm Hg wird nicht empfohlen.
Es wird empfohlen, dass bei einer Änderung der Dosis der systemische Blutdruck und die Herzfrequenz überwacht werden, um beim Auftreten einer Hypotonie oder eines systolischen Blutdrucks von 85 mm Hg oder darunter die Infusion zu stoppen.
Risiken einer starken Dosisreduktion oder eines plötzlichen Abbruchs der Therapie
Wie mit jedem stark wirksamen Vasodilatator kann ein plötzlicher Therapieabbruch oder ein starke Reduktion der Dosierung eine Verschlechterung der pulmonalen arteriellen Hypertonie bewirken, und sollte deshalb vermieden werden.
Risiko von Blutungen
Wie bei dieser häufig mit Antikoagulanzien behandelten Patientengruppe zu erwarten, traten Blutungen häufig auf. Aufgrund seiner Wirkung auf die Thrombozytenaggregation kann Remodulin das Risiko von Blutungen erhöhen. In kontrollierten klinischen Studien wurde eine erhöhte Inzidenz von Epistaxis und gastrointestinalen Blutungen beobachtet (einschliesslich rektalen Blutungen, Zahnfleischblutungen und Blutstuhl). Es gab auch Berichte von Hämoptyse, Hämatemesis und Hämaturie, aber diese traten mit derselben oder einer niedrigeren Frequenz als in der Placebogruppe auf.
Gleichzeitige Anwendung anderer Arzneimittel
Die gleichzeitige Verabreichung eines Cytochrom-P450-(CYP2C8)-Enzyminhibitors (z.B. Gemfibrozil) kann zu einer erhöhten Exposition (sowohl Cmax als auch AUC) gegenüber Treprostinil führen. Bei einer erhöhten Exposition besteht die Wahrscheinlichkeit eines verstärkten Auftretens von unerwünschten Wirkungen in Verbindung mit der Verabreichung von Treprostinil. Daher ist eine Herabsetzung der Dosis in Betracht zu ziehen (siehe «Interaktionen»).
Die gleichzeitige Verabreichung eines CYP2C8-Enzyminduktors (z.B. Rifampicin) kann zu einer verminderten Exposition gegenüber Treprostinil führen. Bei einer verminderten Exposition besteht die Wahrscheinlichkeit einer reduzierten klinischen Wirksamkeit. Daher ist eine höhere Dosierung von Treprostinil in Betracht zu ziehen (siehe «Interaktionen»).
Das Nutzen-/Risikoverhältnis von Remodulin wurde bei pulmonaler arterieller Hypertonie mit einhergehendem Links-Rechts-Shunt, einhergehender portaler Hypertonie oder HIV-Infektion nicht untersucht.
Remodulin enthält max. 75 mg (Dosisstärke 5 mg/ml) Natrium pro 20 ml Durchstechflasche, entsprechend 3.75% der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g. Das muss bei Patienten mit einer kontrollierten Natriumdiät in Betracht gezogen werden.
Interaktionen
In klinischen Studien wurde Remodulin gleichzeitig mit den folgenden Arzneimitteln angewendet: Antikoagulanzien, Diuretika, Herzglykoside, Calciumantagonisten, Analgetika, Antipyretika, nicht-steroidale Antiphlogistika, Kortikosteroide und andere.
Bei der gleichzeitigen Anwendung von Remodulin und Diuretika, Antihypertensiva oder anderen Vasodilatatoren besteht erhöhte Gefahr einer systemischen Hypotonie.
Wenn gleichzeitig andere Thrombozyten-Aggregationshemmer oder Antikoagulanzien verabreicht werden, kann Remodulin das Risiko einer Blutung erhöhen.
Pharmakokinetische Interaktionen
In-vivo-Daten
Acetaminophen: Interaktionsstudien wurden mit Acetaminophen (4 g/Tag) bei gesunden Probanden durchgeführt. Acetaminophen zeigte keine klinisch signifikanten Wirkungen auf die Pharmakokinetik von Treprostinil. Aufgrund von in-vitro-Untersuchungen mit Lebermikrosomen sind auch keine Interaktionen mit Phenprocoumon zu erwarten.
Furosemid: Bei Patienten, die Furosemid erhielten, war die Plasma-Clearance reduziert. Möglicherweise besteht eine Interaktion zwischen Treprostinil und Furosemid, da beide Substanzen während der Metabolisierung an der Carboxylat-Gruppe glukuronisiert werden.
Bosentan: In einer pharmakokinetischen Studie am Menschen, bei der Bosentan (250 mg/Tag) und Treprostinil Diolamin (orale Dosis 2 mg/Tag) verabreicht wurden, waren keine pharmakokinetischen Wechselwirkungen zwischen Treprostinil und Bosentan zu beobachten. Es wurde nicht festgestellt, ob die Sicherheit und Wirksamkeit von Remodulin über den parenteralen (subkutanen oder intravenösen) Verabreichungsweg durch Bosentan verändert werden.
Sildenafil: In einer pharmakokinetischen Studie am Menschen, bei der Sildenafil (60 mg/Tag) und Treprostinil Diolamin (orale Dosis 2 mg/Tag) verabreicht wurden, waren keine pharmakokinetischen Wechselwirkungen zwischen Treprostinil und Sildenafil zu beobachten. Es wurde nicht festgestellt, ob die Sicherheit und Wirksamkeit von Remodulin über den parenteralen (subkutanen oder intravenösen) Verabreichungsweg durch Sildenafil verändert werden.
Pharmakodynamische Interaktionen
Thrombozytenaggregationshemmer, einschliesslich nichtsteroidaler Antirheumatika (NSAR) und Antikoagulanzien
Treprostinil kann die Thrombozytenfunktion hemmen. Die gleichzeitige Gabe von Remodulin und Thrombozytenaggregationshemmern, einschliesslich NSAR, Stickstoffmonoxid-Donatoren oder Antikoagulanzien kann das Risiko einer Blutung erhöhen. Patienten, die Antikoagulanzien erhalten, sollten entsprechend der Empfehlungen der üblichen medizinischen Praxis streng kontrolliert werden. Die gleichzeitige Verabreichung weiterer Thrombozytenaggregationshemmer sollte bei Patienten vermieden werden, die Antikoagulanzien erhalten. Eine subkutane Dauerinfusion von Treprostinil hatte keine Auswirkung auf die Pharmakodynamik und Pharmakokinetik einer einzelnen Dosis Warfarin (25 mg).
Bisher liegen keine klinischen Daten vor, die eine Erhöhung des Blutungsrisikos bei gleichzeitiger Gabe von Treprostinil mit den Stickstoffmonoxid-Donatoren bestätigen.
Enzyminduktoren
Rifampicin: Pharmakokinetische Studien am Menschen mit oralem Treprostinil Diolamin deuteten darauf hin, dass die gleichzeitige Verabreichung des CYP2C8-Enzyminduktors Rifampicin zu einer (um etwa 20-30 %) verminderten Exposition gegenüber Treprostinil führt. Es wurde nicht festgestellt, ob die Sicherheit und Wirksamkeit von Remodulin über den parenteralen (subkutanen oder intravenösen) Verabreichungsweg durch Rifampicin verändert werden. Wird Rifampicin im Anschluss an die Titrierungsphase zur Medikation des Patienten hinzugenommen bzw. weggelassen, ist eine Dosisanpassung von Treprostinil in Betracht zu ziehen.
CYP2C8-Induktoren (z.B. Phenytoin, Carbamazepin, Phenobarbital und Johanniskraut) können zu einer verminderten Exposition gegenüber Treprostinil führen. Wird ein CYP2C8-Induktor im Anschluss an die Titrierungsphase zur Medikation des Patienten hinzugenommen bzw. weggelassen, ist eine Dosisanpassung von Treprostinil in Betracht zu ziehen.
Enzyminhibitoren
Gemfibrozil: Pharmakokinetische Studien am Menschen mit oralem Treprostinil Diolamin zeigen, dass die gleichzeitige Verabreichung des Cytochrom-P450-(CYP2C8)-Enzyminhibitors Gemfibrozil die Exposition (sowohl Cmax als auch AUC) gegenüber Treprostinil verdoppelt. Es wurde nicht festgestellt, ob die Sicherheit und Wirksamkeit von Remodulin über den parenteralen (subkutanen oder intravenösen) Verabreichungsweg durch CYP2C8-Inhibitoren verändert werden. Wird ein CYP2C8-Inhibitor (z.B. Gemfibrozil, Trimethoprim und Deferasirox) im Anschluss an die Titrierungsphase zur Medikation des Patienten hinzugenommen bzw. weggelassen, ist eine Dosisanpassung von Treprostinil in Betracht zu ziehen.
Schwangerschaft/Stillzeit
Schwangerschaft
In Kaninchen fand man in den Föten unter maternaler Toxizität (bei Dosen von 150 und 300 ng/kg/min) eine erhöhte Zahl von Skelettvariationen. Sonst wurden keine adversen Effekte in reproduktionstoxikologischen Studien festgestellt (siehe Präklinische Daten).
Es liegen keine Studien bei schwangeren Frauen vor. Remodulin sollte während der Schwangerschaft nur angewendet werden, wenn es unbedingt notwendig ist.
Stillzeit
Es ist nicht bekannt, ob Treprostinil in die Muttermilch ausgeschieden und vom Kind systemisch aufgenommen wird. Daher sollte während der Stillzeit Remodulin nicht angewendet werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Bei Behandlungsbeginn oder nach Dosisanpassungen können unerwünschte Wirkungen wie symptomatische, systemische Hypotonie oder Schwindel auftreten, die die Verkehrstüchtigkeit und die Sicherheit beim Bedienen von Maschinen beeinträchtigen können. Auf Grund der möglichen unerwünschten Wirkungen ist beim Lenken von Fahrzeugen und beim Bedienen von Maschinen Vorsicht geboten.
Unerwünschte Wirkungen
Eine Interpretation der unerwünschten Wirkungen ist dadurch erschwert, dass einzelne klinische Symptome der pulmonalen arteriellen Hypertonie (z.B. Schwindel, Ohnmacht) ähnlich sind. Unerwünschte Wirkungen, die wahrscheinlich auf die zugrunde liegende Krankheit zurückzuführen sind, schliessen Atemnot, Müdigkeit, Brustschmerzen, rechtsventrikulärer Herzschlag und Blässe ein.
Einige unerwünschte Wirkungen können eindeutig Remodulin zugeschrieben werden. Die häufigsten bei der subkutanen Anwendung sind Reaktionen an der Infusionsstelle (83%) und Schmerzen an der Infusionsstelle (85%), welche aber von den meisten Patienten ertragen werden. Den Reaktionen an der Infusionsstelle wurden alle Ereignisse wie Erytheme, Verhärtungen, Hautausschlag usw. ausser Schmerzen oder Blutungen/blaue Flecken zugeordnet.
Die Häufigkeitsangaben sind wie folgt definiert:
«sehr häufig» (≥1/10)
«häufig» (<1/10, ≥1/100)
«gelegentlich» (<1/100, ≥1/1000)
«selten» (<1/1000, ≥1/10'000)
«sehr selten» (<1/10'000)
«nicht bekannt» (kann aus den verfügbaren Daten nicht abgeschätzt werden)
Infektionen und parasitäre Erkrankungen
Nicht bekannt: Infektion des Blutkreislaufs im Zusammenhang mit dem Zentralvenenkatheter, Sepsis, Bakteriämie**, Infektion an der Infusionsstelle, Abszessbildung an der subkutanen Infusionsstelle, Cellulitis.
Erkrankungen des Blutes und des Lymphsystems
Nicht bekannt: Thrombozytopenie.
Erkrankungen des Nervensystems
Sehr häufig: Kopfschmerzen (30%).
Häufig: Schwindel.
Herzerkrankungen
Nicht bekannt: High-Output-Herzinsuffizienz.
Gefässerkrankungen
Sehr häufig: Vasodilatation/Hitzewallung (Flushing) (11%).
Häufig: Hypotonie, Blutungsereignis§.
Nicht bekannt: Thrombophlebitis*.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Durchfall (22%), Übelkeit (19%).
Häufig: Erbrechen.
Erkrankungen der Haut und des Unterhautgewebes
Sehr häufig: Hautausschlag (12%).
Häufig: Pruritus.
Nicht bekannt: Generalisierte makuläre oder papuläre Ausschläge.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Sehr häufig: Kieferschmerzen (13%).
Häufig: Myalgie, Arthralgie, Gliederschmerzen.
Nicht bekannt: Knochenschmerzen.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Schmerzen an der Infusionsstelle (85%), Reaktionen an der Infusionsstelle (83%), Blutung oder Hämatom (21%).
Häufig: Ödeme.
* Fälle von Thrombophlebitis im Zusammenhang mit peripherer intravenöser Infusion wurden gemeldet.
** Lebensbedrohliche Ereignisse und Todesfälle wurden gemeldet.
§ Siehe Abschnitt «Beschreibung ausgewählter unerwünschter Wirkungen»
Beschreibung ausgewählter Nebenwirkungen
Blutungsereignisse
Blutungen traten häufig auf, was bei einer Patientenpopulation mit einem hohen Anteil an Patienten, die mit Antikoagulanzien behandelt wurden, zu erwarten ist. Aufgrund seiner Auswirkungen auf die Thrombozytenaggregation kann Remodulin das Blutungsrisiko erhöhen, was sich in kontrollierten klinischen Studien durch ein vermehrtes Auftreten von Epistaxis und Magen-Darm-Blutungen (einschliesslich gastrointestinaler Blutungen, rektaler Blutung, Gaumenblutung und Blutstuhl) manifestierte. Ausserdem wurden Hämoptyse, Hämatemesis und Hämaturie gemeldet, aber diese traten mit derselben oder einer niedrigeren Frequenz als in der Placebogruppe auf.
Unerwünschte Ereignisse mit Bezug zur Art der intravenösen Verabreichung
In einer beschränkten Anzahl von klinischen Studien wurde die Verabreichung von Remodulin als intravenöse Infusion mittels Zentralvenenkatheter untersucht. Das Gesamtprofil der unerwünschten Ereignisse in diesen Studien ist vergleichbar mit demjenigen bei der subkutanen Verabreichung von Remodulin. Dies war zu erwarten aufgrund der Bioäquivalenz, die für den intravenösen und subkutanen Verabreichungsweg aufgezeigt werden konnte. Wie jedoch mit jedem für längere Zeit angebrachten Zentralvenenkatheter bestehen gewisse Risiken im Zusammenhang mit dieser Verabreichungsform. Dies beinhaltet Schmerzen an der Eintrittsstelle des Katheters, lokale Infektionen, Sepis, Bildung von Thromben und daraus resultierende Verschlusskrankheit der Gefässe sowie Störungen beim Verabreichungssystem, die in einer unbeabsichtigten Verabreichung eines Bolus resultieren oder in einer unbeabsichtigten Dosisreduktion. Dies kann durch die Über- oder Unterdosierung von Remodulin zu einem Auftreten von entsprechenden Symptomen führen.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Die Zeichen und Symptome der Überdosierung mit Remodulin während der klinischen Studien sind ähnlich den erwarteten dosis-limitierenden pharmakologischen Wirkungen von Remodulin, wie Hitzewallungen (Flushing), Kopfschmerzen, Hypotonie, Übelkeit, Erbrechen und Durchfall. Die meisten Ereignisse waren selbstlimitierend und verschwanden nach einer Dosisreduktion oder nach Abbruch der Behandlung. In vereinzelten Fällen wurde auch eine Entgleisung der pulmonalen arteriellen Hypertonie beobachtet.
Eigenschaften/Wirkungen
ATC-Code
B01AC21
Remodulin ist ein synthetisches Prostazyklin.
Wirkungsmechanismus
Die pharmakologischen Hauptwirkungen von Treprostinil bestehen in der direkten Vasodilatation des systemischen arteriellen und pulmonären Gefässbettes und der Hemmung der Thrombozytenaggregation. Beim Tier führt die gefässerweiternde Wirkung eine Senkung der Nachlast des rechten sowie des linken Ventrikels und eine Erhöhung des Herzschlag- sowie des Systolenvolumens herbei. Die Auswirkung von Treprostinil auf den Herzrhythmus beim Tier hängt von der verabreichten Dosis ab. Es wurde keine nennenswerte Beeinflussung der Herzüberleitung beobachtet.
Pharmakodynamik
Sämtliche Ergebnisse zweier randomisierter, placebokontrollierter, doppelblinder Grundstudien haben gezeigt, dass die durchschnittliche Besserung der folgenden hämodynamischen Parameter nach einer 2-wöchigen Behandlung mit Remodulin in Verbindung mit der klassischen Behandlung in Bezug auf Vergleichswerte ausgeprägter war als mit Placebo in Verbindung mit der klassischen Behandlung: Senkung des maximalen arteriellen Pulmonaldrucks (mAPD), des arteriellen pulmonalen Widerstands (APW), des pulmonalen Gefässwiderstand-Indexes (PVRI) und des systemischen Gefässwiderstand-Indexes (SVRI) sowie Erhöhung der inspiratorischen Kapazität und der venösen Sauerstoffsättigung.
Klinische Wirksamkeit
Die Bewegungsfähigkeit von Patienten wurde mit Hilfe eines Gehtest sechs Minuten lang gemessen; sie war bei Patienten, die 12 Monate lang zusammen mit einer klassischen Behandlung eine subkutan verabreichte Infusion von Remodulin erhalten hatten (n= 232), besser (Median 10 m versus 0 m) als bei Probanden unter Placebo in Verbindung mit der klassischen Behandlung (n= 236) (p= 0,0064). Die Besserung war bereits nach 6 Wochen Behandlung sichtbar. Die bessere Bewegungsfähigkeit war mit einer statistisch signifikanten Besserung der Dyspnoe und Müdigkeit, die nach der Dyspnoe-Müdigkeit-Skala von Borg bewertet worden war, sowie einer pulmonalen arteriellen Hypertension verbunden.
Pharmakokinetik
Absorption
Wird Remodulin subkutan infundiert, werden die Steady-State-Plasmakonzentrationen normalerweise 15 bis 18 Stunden nach Beginn der Therapie erreicht und sind bei Infusionsraten von 2.5 bis 125 ng/kg/min proportional zur Dosis. Remodulin wird als subkutane Infusion vollständig resorbiert.
Distribution
Bei gesunden Probanden erreichten die Plasmakonzentrationen nach subkutaner Verabreichung um 1 Uhr nachts und um 10 Uhr morgens ein Maximum sowie um 7 Uhr morgens und 4 Uhr nachmittags ein Minimum. Die Maxima lagen etwa 20 bis 30% höher als die Minima. Eine Dosisanpassung aufgrund der Tagesschwankungen wird nicht als nötig erachtet.
Das mittlere Verteilungsvolumen liegt bei 1.1 l/kg und die Plasma-Clearance bei 589 ml/kg/h. Die subkutane und intravenöse Verabreichung von Remodulin zeigte eine Bioäquivalenz im Steady state in einer Dosis von 10 ng/kg/min.
Metabolismus
Remodulin wird zu einem grossen Teil hepatisch metabolisiert. An der Metabolisierung von Treprostinil ist hauptsächlich CYP2C8 und zu einem geringen Anteil CYP2C9 beteiligt. Auf Grund von in vitro-Daten mit humanen P450 hemmt Remodulin CYP1A2, 2C9, 2C19, 2D6, 2E1 und 3A nicht. Ob Remodulin P450 Isoenzyme induziert, ist derzeit am Menschen nicht untersucht.
Es wurde kein einzelner Hauptmetabolit von Treprostinil beobachtet. Im Urin wurden 5 Metaboliten in einer Menge von 10 bis 15% der verabreichten Dosis gefunden, insgesamt 64%. 3 der Metaboliten sind Oxidationsprodukte der 3-Hydroxyloctyl-Seitenkette, einer ist ein Glucuronid-Konjugat und einer ist nicht identifiziert. Nur 3.7% wurden unverändert im Urin gefunden.
Elimination
Bei subkutaner Applikation zeigt Treprostinil eine scheinbare Eliminationshalbwertszeit von 1.3 h verglichen mit 45 min bei intravenöser Verabreichung.
In einer Studie bei gesunden Probanden mit 14C-Treprostinil wurden über einen Zeitraum von 224 h 78.6% der verabreichten Dosis im Urin und 13.4% in den Faeces gefunden.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Die Plasma-Treprostinil-Exposition (Fläche unter der Plasmakonzentrationszeitkurve; AUC) war bei milder bis moderate Leberfunktionsstörung, Child-Pugh-Klasse A bzw. B, um 260% bis 510% erhöht. Die Plasma-Clearance war bei Patienten mit leichter bis mittlerer Leberfunktionsstörung um bis zu 80% reduziert verglichen mit gesunden Probanden (siehe Dosierung/Anwendung).
Nierenfunktionsstörungen
Es liegen keine speziellen Studien vor. Eine multivariante Analyse zeigte, dass die Plasma-Clearance von Treprostinil mit dem Serum-Kreatinin assoziiert ist.
Ältere Patienten
Bei älteren Patienten wurde eine geringe Reduktion der Steady-State-Clearance (ca. 3%) festgestellt, welche jedoch kaum eine Dosisanpassung rechtfertigt.
Kinder und Jugendliche
Es wurden keine Studien bei Kindern und Jugendlichen durchgeführt, weshalb die Anwendung nicht empfohlen wird.
Anwendung bei Patienten mit Übergewicht
Patienten mit Übergewicht (BMI >30 kg/m²) zeigen eine geringere Clearance von Treprostinil.
Präklinische Daten
Langzeittoxizität (bzw. Toxizität bei wiederholter Verabreichung)
In Studien an Ratten und Kaninchen, die über 26 Wochen durchgeführt worden waren, erwies sich Treprostinil in der Dosierung von 450 ng/kg/Min. bzw. 200 ng/kg/Min. als nicht toxisch.
Mutagenität
Aus den Mutagenitätstests in vitro und in vivo ging weder eine mutagene noch eine klastogene Wirkung von Treprostinil hervor.
Karzinogenität
Es wurden keine Langzeitstudien durchgeführt, um die karzinogene Wirkung zu bewerten.
Reproduktionstoxizität
In Ratten wurden bei Dosen bis zu 900 ng/kg/min und in Kaninchen bis 50 ng/kg/min keine reproduktionstoxikologischen Effekte beobachtet. In Kaninchen fand man bei Dosen von 150 und 300 ng/kg/min und unter maternaler Toxizität eine erhöhte Zahl von Skelettvariationen. Die männliche und die weibliche Reproduktionsfähigkeit war in Ratten bei Dosen bis 450 ng/kg/min nicht beeinflusst.
Sonstige Hinweise
Inkompatibilitäten
Da keine Verträglichkeitsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden, ausser mit Wasser für Injektionszwecke oder mit 0.9% wässriger NaCl-Lösung (siehe Hinweise für die Handhabung).
Haltbarkeit
Ungeöffnete Durchstechflaschen dürfen nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Einmal angestochene Durchstechflaschen dürfen höchstens während 30 Tagen verwendet werden.
Subkutane Anwendung
Das Infusionsbesteck (inkl. Lösungsreservoir) ist alle 48 bis 72 h auszuwechseln.
Intravenöse Anwendung
Die verdünnte Lösung muss zumindest alle 48 Stunden gewechselt werden.
Besondere Lagerungshinweise
Bei Raumtemperatur (15-25 °C) ausserhalb der Reichweite von Kindern lagern.
Hinweise für die Handhabung
Für die intravenöse Verabreichung muss Remodulin mit Wasser für Injektionszwecke oder 0.9% wässriger NaCl-Lösung verdünnt werden. Die Kompatibilität ist in einem Konzentrationsbereich von 0.004 mg/ml bis 0.13 mg/ml belegt.
Bezüglich Anwendung der Pumpe vergleiche «Dosierung/Anwendung».
Es ist zu berücksichtigen, dass sich Patienten mit Langzeit-Zentralvenenkatheter mit Gram-negativen und Gram-positiven Keimen infizieren können. Die Verwendung eines 0.2 Mikrometer in-line Filters wird empfohlen, um das Risiko einer systemischen Infektion als Folge einer Verunreinigung in der Zubereitung des Arzneimittels zu minimieren.
Zulassungsnummer
56120 (Swissmedic).
Zulassungsinhaberin
Gebro Pharma AG, 4410 Liestal.
Stand der Information
Juni 2020.
Dosierungshilfe für die s.c. Applikation
(Berechnungsformel siehe Dosierung/Anwendung)




Dosierungshilfe für die i.v.Apllikation
(Berechnungsformel siehe Dosierung/Anwendung)




Composizione
Principi attivi
Treprostinil (sotto forma di treprostinil sodico).
Sostanze ausiliarie
Sodio citrato diidrato, acido cloridrico, metacresolo, idrossido di sodio, sodio cloruro, acqua per preparazioni iniettabili.
Contenuto totale di sodio per mL:
Forza di dosaggio | 1.0 | 2.5 | 5.0 | 10.0 |
Contenuto totale di sodio (mg) | 3.7 | 3.75 | 3.92 | 3.74 |
Forma farmaceutica e quantità di principio attivo per unità
Soluzione per infusione per uso sottocutaneo o endovenoso:
Flaconcini da 1 mg di treprostinil/ml, da 2,5 mg di treprostinil/ml, da 5 mg di treprostinil/ml e da 10 mg di treprostinil/ml
Indicazioni/Possibilità d'impiego
Trattamento a lungo termine dell'ipertensione polmonare primaria e dell'ipertensione arteriosa polmonare con malattia del tessuto connettivo nei pazienti classificati come classe funzionale NYHA III e IV (secondo la classificazione della New York Heart Association).
Posologia/Impiego
Il trattamento con Remodulin è iniziato in ospedale sotto controllo medico. Remodulin è somministrato per infusione sottocutanea o endovenosa continua. A causa dei rischi correlati con l'utilizzo del catetere venoso centrale permanente, incluse le forme severe di batteriemia e sepsi, che possono essere fatali, l'infusione sottocutanea (non diluita) è la modalità di somministrazione di elezione.
Avvio della terapia
Dose iniziale
La velocità di infusione iniziale raccomandata è di 1,25 ng/kg/min. Se questa dose non è tollerata dal paziente, la velocità di infusione può essere ridotta a 0,625 ng/kg/min.
ATTENZIONE: la soluzione per infusione endovenosa non deve mai essere somministrata non diluita.
Adeguamento della dose/titolazione
L'obiettivo degli aggiustamenti del dosaggio cronico è stabilire una dose alla quale i sintomi dell'ipertensione arteriosa polmonare migliorano, mantenendo nel contempo un profilo degli effetti collaterali tollerabile. La velocità di infusione deve essere aumentata con incrementi di 1,25 ng/kg/min alla settimana per le prime 4 settimane di trattamento e successivamente di 2,5 ng/kg/min alla settimana.
Gli effetti indesiderati dose-dipendenti quali rossore (flushing), cefalea, ipotensione, nausea, vomito e diarrea possono rendere necessario ridurre la velocità di infusione, sebbene possano scomparire anche senza un aggiustamento della dose. Nel caso un effetto indesiderato peggiori e/o diventi intollerabile, la velocità di infusione deve essere ridotta.
Evitare l'interruzione brusca della terapia o una marcata riduzione della dose (vedere «Avvertenze e misure precauzionali»).
Nella maggior parte dei casi, dopo un'interruzione di poche ore, la ripresa dell'infusione può avvenire utilizzando la stessa velocità di somministrazione. Interruzioni per periodi più lunghi possono richiedere una nuova titolazione della dose.
Durante gli studi a lungo termine, le dosi medie raggiunte sono state di 26 ng/kg/min dopo 12 mesi, di 36 ng/kg/min dopo 24 mesi e di 42 ng/kg/min dopo 36 mesi.
Terapia di mantenimento
Infusione continua sottocutanea
La velocità di infusione sottocutanea è calcolata utilizzando la seguente formula:
Velocità di infusione (ml/h):

* Fattore di conversione (0,00006): 60 min/h × 0,000001 mg/ng
Infusione endovenosa continua
Remodulin diluito è somministrato per infusione endovenosa continua attraverso un catetere venoso centrale utilizzando una pompa per infusione.
Può essere anche somministrato temporaneamente attraverso una cannula venosa periferica posizionata preferibilmente in una grande vena. L'uso dell'infusione periferica per più di qualche ora può essere associato ad un aumento del rischio di tromboflebite (vedere «Effetti indesiderati»).
Si raccomanda l'uso di un filtro in linea di 0,2 micrometri per ridurre al minimo il rischio di infezione sistemica causata da contaminazione durante la preparazione del medicamento.
Calcolo della concentrazione richiesta per la soluzione di Remodulin diluita:
Concentrazione finale della soluzione di Remodulin diluita (mg/ml):

Calcolo della quantità di Remodulin richiesta, da diluire con un volume di diluente sufficiente (acqua sterile per preparazioni iniettabili o soluzione di NaCl allo 0,9% per infusioni):
Volume di Remodulin da utilizzare (ml):

La quantità calcolata di Remodulin viene quindi aggiunta al serbatoio e diluita con un volume sufficiente di acqua sterile o soluzione di NaCl allo 0,9%.
Istruzioni posologiche speciali
Pazienti con disturbi della funzionalità epatica
L'esposizione plasmatica a treprostinil (area sottesa alla curva concentrazione plasmatica - tempo; AUC) aumenta del 260%-510% in caso di disturbi della funzionalità epatica da lievi a moderati, rispettivamente di classe A e B di Child-Pugh. La clearance plasmatica di treprostinil è risultata ridotta in misura fino all'80% nei soggetti che presentavano disturbi della funzionalità epatica da lievi a moderati rispetto ai soggetti sani. Si consiglia pertanto di prestare attenzione nel trattamento con Remodulin di pazienti con disturbi della funzionalità epatica e di monitorare attentamente i pazienti per una possibile comparsa di effetti indesiderati legati all'incremento dei livelli di Remodulin (vedere «Farmacocinetica»).
La dose iniziale di Remodulin deve essere ridotta a 0,625 ng/kg/min e gli incrementi devono essere effettuati con cautela.
Non sono disponibili dati di pazienti con severi disturbi della funzionalità epatica.
Pazienti con disturbi della funzionalità renale
Non sono stati condotti studi su pazienti con disturbi della funzionalità renale. Poiché il treprostinil e i suoi metaboliti sono escreti principalmente attraverso le vie urinarie, si raccomanda di prestare attenzione quando si trattano pazienti con disturbi della funzionalità renale. L'aumento della dose deve avvenire con particolare cautela per questi pazienti (vedere «Farmacocinetica»).
Impiego in caso di sovrappeso
I pazienti in sovrappeso (IMC >30 kg/m²) presentano una clearance inferiore di treprostinil.
Pazienti anziani
Non sono disponibili dati sufficienti per i pazienti anziani (≥65 anni). La dose per un paziente anziano deve essere selezionata con cautela, prendendo in considerazione la maggiore frequenza di ridotta funzionalità epatica, renale o cardiaca e la presenza di malattie concomitanti o altre terapie farmacologiche.
Bambini e adolescenti
I dati disponibili su bambini e adolescenti di età inferiore ai 16 anni non sono sufficienti per stabilire se lo schema posologico raccomandato per gli adulti possa essere utilizzato per questo gruppo di pazienti.
Modo di somministrazione
Remodulin è somministrato per infusione sottocutanea o endovenosa continua. A causa dei rischi correlati con l'utilizzo del catetere venoso centrale permanente, incluse le forme severe di batteriemia e sepsi, che possono essere fatali, l'infusione sottocutanea (non diluita) è la modalità di somministrazione di elezione.
Con la somministrazione per infusione sottocutanea, la cannula viene inserita sotto pelle nell'addome o sulla coscia e fissata. Nella somministrazione sottocutanea, il flaconcino di Remodulin è parte della pompa. Il set d'infusione e il relativo serbatoio per il medicamento devono essere sostituiti ogni 48-72 ore.
Con la somministrazione per infusione endovenosa viene utilizzato un catetere centrale permanente. Remodulin viene somministrato con un flusso continuo e costante mediante una piccola pompa fissata all'altezza della vita, sul polpaccio, sulla coscia o sull'addome. Per la somministrazione endovenosa, Remodulin è utilizzato diluito. La soluzione viene portata all'ago mediante un tubicino in polivinile.
Per l'infusione endovenosa, la soluzione diluita deve essere sostituita almeno ogni 48 ore
(in virtù della stabilità di Remodulin).
Il personale dell'ospedale illustrerà ai pazienti nel dettaglio come utilizzare la pompa, come collegare il set d'infusione, come sostituire il serbatoio di Remodulin e come eseguire l'infusione continua. Rispettare le norme igieniche di manipolazione.
La velocità di flusso di Remodulin viene regolata settimanalmente. Generalmente la dose viene leggermente aumentata a seconda dei sintomi dell'ipertensione polmonare. Nel caso in cui si manifestino effetti collaterali intollerabili, la dose viene aggiustata secondo le indicazioni del medico.
L'utilizzo corretto della pompa per infusione è descritto nelle relative istruzioni per l'uso.
È importante utilizzare la concentrazione corretta di Remodulin e il set per infusione giusto.
Un malfunzionamento della pompa o un'occlusione del set per infusione possono provocare il peggioramento dei sintomi dell'ipertensione arteriosa polmonare. In caso di interruzione dell'erogazione di Remodulin, seguire le istruzioni per l'uso della pompa. Il paziente deve avere sempre accesso ad una pompa di infusione, a un set per infusione e a scorte di Remodulin di riserva.
La pompa di infusione utilizzata per somministrare Remodulin dovrà essere:
- piccola e leggera,
- in grado di regolare la velocità di infusione con incrementi di 0,002 ml/h per l'infusione sottocutanea e di circa 0,05 ml/h per l'infusione endovenosa. La velocità di infusione tipica delle pompe per infusione endovenosa è compresa tra 0,4 e 2 ml all'ora.
- dotata di allarmi di occlusione/interruzione del flusso, batteria in esaurimento, errore di programmazione e malfunzionamento del motore,
- con accuratezza del flusso di ±6% o superiore,
- guidata da pressione positiva,
- il serbatoio deve essere in polivinile cloruro, polipropilene o vetro.
Remodulin è disponibile in concentrazioni da 1 mg/ml, 2,5 mg/ml, 5 mg/ml e 10 mg/ml.
Le tabelle per la regolazione del dosaggio sono riportate in calce all'informazione professionale.
Controindicazioni
- Ipersensibilità a uno dei componenti di Remodulin o agli analoghi strutturali di treprostinil,
- Ipertensione arteriosa polmonare correlata a sindrome veno-occlusiva,
- Insufficienza cardiaca congestizia dovuta a severa disfunzione del ventricolo sinistro,
- Disturbi severi della funzionalità epatica (classe C di Child-Pugh),
- Ulcera gastrointestinale attiva, emorragia intracranica, ferite o altre condizioni di sanguinamento,
- Difetti valvolari congeniti o acquisiti con disturbi della funzionalità miocardica clinicamente rilevanti non legati all'ipertensione polmonare,
- Cardiopatia coronarica severa o angina instabile; infarto miocardico negli ultimi sei mesi; insufficienza cardiaca scompensata se non sotto la stretta supervisione medica; aritmie severe; eventi cerebrovascolari (ad es. attacco ischemico transitorio, ictus) negli ultimi tre mesi.
Avvertenze e misure precauzionali
Misure precauzionali generali
Remodulin deve essere utilizzato unicamente da medici specialisti con esperienza nella diagnosi e nel trattamento dell'ipertensione arteriosa polmonare. La decisione di iniziare il trattamento con Remodulin deve tenere in considerazione l'elevata probabilità che la terapia debba protrarsi per un periodo prolungato, probabilmente per diversi anni. Deve essere pertanto attentamente valutata la capacità del paziente di somministrare Remodulin e di farsi carico della manutenzione del sistema di infusione.
Remodulin deve essere somministrato solo per via sottocutanea o diluito per via endovenosa.
Remodulin è un potente vasodilatatore polmonare e sistemico. Il trattamento con Remodulin deve pertanto iniziare con personale adeguatamente qualificato e attrezzato per il monitoraggio fisiologico e il trattamento di emergenza. Nel corso degli studi clinici sono stati intrapresi aggiustamenti della dose a causa dei sintomi dell'ipertensione arteriosa polmonare e degli effetti collaterali di Remodulin.
Rischi associati alla somministrazione di Remodulin per via endovenosa
Batteriemie associate al catetere venoso centrale e sepsi dall'esito potenzialmente letale sono state riportate in pazienti trattati con Remodulin per infusione endovenosa. Tali rischi sono riconducibili alla via di somministrazione del medicamento. L'infusione continua sottocutanea del medicamento (non diluito) è pertanto il modo di somministrazione di elezione. L'infusione continua per via endovenosa deve essere riservata a pazienti stabilizzati con l'infusione sottocutanea di treprostinil che diventano intolleranti alla via sottocutanea e in cui questi rischi sono considerati accettabili.
Adottare tecniche asettiche per la preparazione e somministrazione di Remodulin.
Rischio di ipotensione sistemica
In pazienti con bassa pressione arteriosa sistemica, il trattamento con treprostinil può aumentare il rischio di ipotensione sistemica. Il trattamento non è raccomandato per pazienti con pressione arteriosa sistolica inferiore a 85 mm Hg.
Si raccomanda di monitorare la pressione arteriosa sistemica e la frequenza cardiaca durante i cambiamenti di dose, interrompendo l'infusione qualora insorgano sintomi di ipotensione o si rilevi una pressione arteriosa sistolica di 85 mm Hg o inferiore.
Rischi associati alla riduzione molto marcata della dose o a un'improvvisa interruzione del trattamento
Come per qualunque altro potente vasodilatatore, la brusca interruzione del trattamento o una marcata riduzione della dose possono causare il peggioramento dell'ipertensione arteriosa polmonare e devono pertanto essere evitate.
Rischio di sanguinamento
Casi di sanguinamento si sono rivelati comuni, come era prevedibile per questo gruppo di pazienti spesso trattati con anticoagulanti. A causa degli effetti sull'aggregazione piastrinica, Remodulin può aumentare il rischio di sanguinamento. In studi clinici controllati è stato osservato un aumento dell'incidenza di epistassi e sanguinamento gastrointestinale (comprese emorragia rettale, emorragia gengivale ed ematochezia). Sono stati riportati anche casi di emottisi, ematemesi ed ematuria, ma questi si sono verificati con frequenza pari o inferiore rispetto al gruppo placebo.
Uso concomitante di altri medicamenti
La somministrazione concomitante di un inibitore degli enzimi del citocromo P450 (CYP2C8) (per es. gemfibrozil) può aumentare l'esposizione (sia la Cmax che l'AUC) a treprostinil. È probabile che una maggiore esposizione porti all'aumento degli effetti indesiderati associati con la somministrazione di treprostinil. Si deve pertanto considerare una riduzione della dose (vedere «Interazioni»).
La somministrazione concomitante di un induttore dell'enzima CYP2C8 (per es. rifampicina) può ridurre l'esposizione a treprostinil. È probabile che un'esposizione minore riduca l'efficacia clinica. Si deve considerare pertanto un aumento della dose di treprostinil (vedere «Interazioni»).
Il rapporto rischi/benefici di Remodulin non è stato studiato nell'ipertensione arteriosa polmonare associata a shunt cardiaco sinistro-destro, ipertensione portale o infezione da HIV.
Remodulin contiene fino a un massimo di 75 mg (concentrazione 5 mg/ml) di sodio per flaconcino da 20 ml, corrispondenti al 3.75% dell'apporto giornaliero massimo di 2 g di sodio con la dieta raccomandato dall'OMS per gli adulti. Si tratta di un dato da tenere in considerazione in pazienti che seguono una dieta a basso contenuto di sodio.
Interazioni
La somministrazione concomitante di Remodulin con diuretici, agenti antipertensivi o altri vasodilatatori aumenta il rischio di ipotensione sistemica.
La somministrazione concomitante di Remodulin e inibitori dell'aggregazione piastrinica o anticoagulanti può aumentare il rischio di sanguinamento.
Negli studi clinici Remodulin è stato somministrato in concomitanza con i seguenti medicamenti: anticoagulanti, diuretici, glicosidi cardiaci, calcio-antagonisti, analgesici, antipiretici, antinfiammatori non steroidei, corticosteroidi e altri.
Interazioni farmacocinetiche
Dati in vivo
Acetaminofene: Studi di interazione con acetaminofene (4 g/die) sono stati condotti su soggetti sani. L'acetaminofene non ha evidenziato effetti clinicamente rilevanti sulla farmacocinetica di treprostinil. Alla luce degli studi in vitro condotti con microsomi epatici non sono prevedibili interazioni neppure con fenprocumone.
Furosemide: La clearance plasmatica è risultata ridotta in pazienti trattati con furosemide. Sussiste probabilmente un'interazione tra treprostinil e furosemide, poiché entrambe le sostanze vengono glucuronizzate nel gruppo carbossilato durante il metabolismo.
Bosentan: In uno studio di farmacocinetica nell'uomo condotto con bosentan (250 mg/die) e treprostinil diolamina (dose orale di 2 mg/die), non sono state osservate interazioni farmacocinetiche fra treprostinil e bosentan. Non è stato stabilito se la sicurezza e l'efficacia di Remodulin somministrato per via parenterale (sottocutanea o endovenosa) vengano alterate dal bosentan.
Sildenafil: In uno studio di farmacocinetica nell'uomo condotto con sildenafil (60 mg/die) e treprostinil diolamina (dose orale di 2 mg/die), non sono state osservate interazioni farmacocinetiche fra treprostinil e sildenafil. Non è stato stabilito se la sicurezza e l'efficacia di Remodulin somministrato per via parenterale (sottocutanea o endovenosa) vengano alterate dal sildenafil.
Interazioni farmacodinamiche
Inibitori dell'aggregazione piastrinica, inclusi antireumatici non steroidei (FANS) e anticoagulanti
Il treprostinil può inibire la funzione piastrinica. La somministrazione concomitante di Remodulin e inibitori dell'aggregazione piastrinica, compresi FANS, donatori di ossido nitrico o anticoagulanti può aumentare il rischio di sanguinamento. La stretta sorveglianza dei pazienti che assumono anticoagulanti deve essere mantenuta in conformità alle comuni raccomandazioni della pratica medica. L'uso concomitante di altri antiaggreganti piastrinici deve essere evitato nei pazienti che assumono anticoagulanti. L'infusione sottocutanea continua di treprostinil non ha avuto alcun effetto sulla farmacodinamica e farmacocinetica di una singola dose (25 mg) di warfarin.
Non sono attualmente disponibili dati clinici che confermino un maggiore rischio di sanguinamento in caso di prescrizione del treprostinil assieme a donatori di ossido nitrico.
Induttori degli enzimi
Rifampicina: Studi di farmacocinetica nell'uomo con treprostinil diolamina orale hanno indicato che la somministrazione concomitante di rifampicina, un induttore dell'enzima CYP2C8, riduce l'esposizione a treprostinil (del 20-30% circa). Non è stato stabilito se la sicurezza e l'efficacia di Remodulin somministrato per via parenterale (sottocutanea o endovenosa) vengano alterate dalla rifampicina. Se, dopo il periodo di titolazione, viene aggiunta o sottratta rifampicina ai medicamenti del paziente, si deve considerare un aggiustamento della dose di treprostinil.
Gli induttori del CYP2C8 (per es. fenitoina, carbamazepina, fenobarbital e iperico) possono ridurre l'esposizione a treprostinil. Se, dopo il periodo di titolazione, viene aggiunto o sottratto un induttore del CYP2C8 ai medicamenti del paziente, si deve considerare un aggiustamento della dose di treprostinil.
Inibitori degli enzimi
Gemfibrozil: Studi di farmacocinetica nell'uomo con treprostinil diolamina orale indicano che la somministrazione concomitante di gemfibrozil, un inibitore degli enzimi del citocromo P450 (CYP2C8), raddoppia l'esposizione a treprostinil (sia la Cmax che l'AUC). Non è stato stabilito se la sicurezza e l'efficacia di Remodulin somministrato per via parenterale (sottocutanea o endovenosa) vengano alterate dagli inibitori del CYP2C8. Se, dopo il periodo di titolazione, viene aggiunto o sottratto un inibitore del CYP2C8 (per es. gemfibrozil, trimetoprim e deferasirox) ai medicamenti del paziente, si deve considerare un aggiustamento della dose di treprostinil.
Gravidanza/Allattamento
Gravidanza
Nel coniglio, i feti hanno evidenziato un aumento del numero di variazioni scheletriche in presenza di tossicità materna (a dosi di 150 e 300 ng/kg/min). Gli studi di tossicità riproduttiva non hanno evidenziato altri effetti avversi.
Non sono disponibili studi su donne in gravidanza. Remodulin va utilizzato durante la gravidanza solo se strettamente necessario.
Allattamento
Non è noto se il treprostinil sia escreto nel latte materno e se sia assorbito a livello sistemico dal lattante. Remodulin non deve pertanto essere impiegato durante l'allattamento.
Effetti sulla capacità di condurre veicoli e sull'impiego di macchine
L'inizio del trattamento o gli aggiustamenti del dosaggio possono essere accompagnati da effetti indesiderati quali ipotensione sistemica sintomatica o capogiri che possono compromettere la capacità di guidare veicoli e azionare macchinari. Alla luce dei possibili effetti indesiderati, si raccomanda prudenza nella guida di veicoli e nell'utilizzo di macchinari.
Effetti indesiderati
L'interpretazione degli effetti indesiderati è complicata dal fatto che alcuni sintomi clinici dell'ipertensione arteriosa polmonare (per es. capogiri, perdita di coscienza) sono simili.
Gli effetti indesiderati probabilmente associati alla patologia sottostante includono difficoltà respiratorie, stanchezza, dolore al petto, battito ventricolare destro e pallore.
Alcuni effetti indesiderati sono chiaramente attribuibili a Remodulin. Gli effetti più comuni in caso di somministrazione sottocutanea sono reazioni nel sito d'infusione (83%) e dolore nel sito d'infusione (85%), tollerati tuttavia dalla maggioranza dei pazienti. Le reazioni del sito d'infusione includono tutti gli eventi quali eritemi, indurimento, eruzione cutanea, ecc., fatta eccezione per dolore o sanguinamenti/ematomi.
Gli effetti indesiderati devono essere classificati secondo la classificazione sistemica organica MedDRA e la frequenza secondo la seguente convenzione:
«molto comune» (≥1/10)
«comune» (≥1/100, <1/10),
«non comune» (≥1/1000, <1/100)
«raro» (≥1/10000, <1/1000)
«molto raro» (<1/10 000)
«non nota» (la frequenza non può essere definita sulla base dei dati disponibili)
Infezioni ed infestazioni
Non nota: infezione del flusso sanguigno associata al catetere venoso centrale, sepsi, batteriemia**, infezione nella sede di infusione, formazione di ascesso nella sede di infusione sottocutanea, cellulite.
Patologie del sistema emolinfopoietico
Non nota: trombocitopenia.
Patologie del sistema nervoso
Molto comune: cefalea (30%).
Comune: capogiri.
Patologie cardiache
Non nota: insufficienza cardiaca ad alta gittata.
Patologie vascolari
Molto comune: vasodilatazione/vampate (flushing) (11%).
Comune: ipotensione, eventi emorragici§.
Non nota: tromboflebite*.
Patologie gastrointestinali
Molto comune: diarrea (22%), nausea (19%).
Comune: vomito.
Patologie della cute e del tessuto sottocutaneo
Molto comune: eruzione cutanea (12%).
Comune: prurito.
Non nota: eruzioni maculari o papulari generalizzate.
Patologie del sistema muscoloscheletrico e del tessuto connettivo
Molto comune: dolore alla mandibola (13%).
Comune: mialgia, artralgia, dolore agli arti.
Non nota: dolore osseo.
Patologie generali e condizioni relative alla sede di somministrazione
Molto comune: dolore nella sede di infusione (85%), reazioni nella sede di infusione (83%), sanguinamento o ematoma (21%).
Comune: edemi.
*Sono stati riferiti casi di tromboflebite associata all'infusione endovenosa periferica.
**Sono stati riferiti casi potenzialmente letali e letali.
§ Vedere paragrafo «Descrizione di effetti indesiderati selezionati»
Descrizione di effetti indesiderati selezionati
Eventi emorragici
Casi di sanguinamento si sono rivelati comuni, come era prevedibile per una popolazione con un'alta percentuale di pazienti trattati con anticoagulanti. A causa degli effetti sull'aggregazione piastrinica, Remodulin può aumentare il rischio di sanguinamento, come dimostrato da un aumento dell'incidenza di epistassi e sanguinamento gastrointestinale (comprese emorragia gastrointestinale, emorragia rettale, emorragia gengivale ed ematochezia) in studi clinici controllati. Sono stati riportati anche casi di emottisi, ematemesi ed ematuria, ma questi si sono verificati con frequenza pari o inferiore rispetto al gruppo placebo.
Eventi indesiderati attribuibili alla via di somministrazione endovenosa
In un numero ridotto di studi clinici è stata esaminata la somministrazione di Remodulin per infusione endovenosa mediante catetere venoso centrale. Il profilo complessivo degli eventi indesiderati riportati in questi studi è analogo a quello della somministrazione di Remodulin per via sottocutanea. Il risultato era prevedibile alla luce della bioequivalenza dimostrata per le vie di somministrazione sottocutanea ed endovenosa. Come sempre nel caso di utilizzo prolungato di un catetere venoso centrale, vi sono tuttavia rischi attribuibili a questo sistema di erogazione del medicamento. Tali rischi includono dolore nella sede di inserimento del catetere, infezioni locali, sepsi, formazione di trombi e conseguente vasculopatia ostruttiva nonché malfunzionamenti del sistema di erogazione che possono causare la somministrazione accidentale di un bolo o la riduzione accidentale della dose. Si possono in questo caso manifestare sintomi correlati al sovradosaggio o sottodosaggio di Remodulin.
La notifica di effetti collaterali sospetti dopo l'omologazione del medicamento è molto importante. Consente una sorveglianza continua del rapporto rischio-benefico del medicamento. Chi esercita una professione sanitaria è invitato a segnalare qualsiasi effetto indesiderato sospetto, nuovo o serio, attraverso il portale online ElViS (Electronic Vigilance System). Maggiori informazioni sul sito www.swissmedic.ch.
Posologia eccessiva
I segni e sintomi di sovradosaggio di Remodulin osservati durante gli studi clinici sono simili agli attesi effetti farmacologici limitanti la dose di Remodulin, quali vampate (flushing), cefalea, ipotensione, nausea, vomito e diarrea. La maggioranza degli eventi era di natura autolimitante ed è scomparsa dopo la riduzione della dose o l'interruzione del trattamento. In casi isolati è stato riferita anche ipertensione arteriosa polmonare scompensata.
Proprietà/Effetti
Codice ATC
B01AC21
Remodulin è una prostaciclina sintetica.
Meccanismo d'azione
I principali effetti farmacologici di treprostinil consistono nell'effetto diretto di vasodilatazione sulla circolazione arteriosa sistemica e polmonare e nell'inibizione dell'aggregazione piastrinica. Negli animali, gli effetti vasodilatatori riducono il postcarico ventricolare destro e sinistro e aumentano la gittata cardiaca e la gittata sistolica. L'effetto del treprostinil sul ritmo cardiaco negli animali varia in base al dosaggio. Non è stato osservato alcun effetto importante sulla conduzione cardiaca.
Farmacodinamica
Tutti i risultati di due studi di base randomizzati, in doppio cieco, controllati verso placebo, hanno evidenziato che il miglioramento medio dei seguenti parametri emodinamici rispetto ai valori di base dopo un trattamento di 2 settimane con Remodulin in associazione con il trattamento classico era maggiore rispetto a quello ottenuto con il placebo in associazione con il trattamento classico: diminuzione della pressione arteriosa polmonare massima (mAPD), della resistenza delle arterie polmonari (APW), dell'indice di resistenza vascolare polmonare (PVRI) e dell'indice di resistenza vascolare sistemica (SVRI) nonché aumento della capacità inspiratoria e della saturazione venosa di ossigeno.
Efficacia clinica
La capacità di esercizio fisico dei pazienti è stata esaminata con il test del cammino di sei minuti; nei pazienti che hanno ricevuto per 12 mesi un'infusione sottocutanea di Remodulin in associazione al trattamento classico (n= 232), detta capacità è risultata migliore (mediana 10 m rispetto a 0 m) rispetto ai soggetti trattati con placebo in associazione al trattamento classico (n= 236) (p= 0,0064). Il miglioramento è risultato evidente già dopo 6 settimane di trattamento. La maggiore capacità di esercizio fisico era associata a un miglioramento statisticamente significativo della dispnea e dello sforzo, valutata sulla scala di Borg per la dispnea e la percezione dello sforzo, nonché dell'ipertensione arteriosa polmonare.
Farmacocinetica
Assorbimento
Le concentrazioni plasmatiche allo stato stazionario sono di solito raggiunte 15 - 18 ore dall'inizio dell'infusione di Remodulin per via sottocutanea e sono proporzionali alla dose a velocità di infusione da 2,5 fino a 125 ng/kg/min. Remodulin somministrato come infusione sottocutanea è completamente assorbito.
Distribuzione
Le concentrazioni plasmatiche in soggetti sani hanno raggiunto il livello massimo dopo la somministrazione sottocutanea all'una di notte e alle 10 del mattino e toccato il livello minimo alle 7 del mattino e alle 4 del pomeriggio. I valori massimi sono risultati superiori al minimo di circa il 20%-30%. L'aggiustamento della dose dovuto alle oscillazioni giornaliere non è considerato necessario.
Il volume medio di distribuzione è di 1,1 l/kg e la clearance plasmatica è di 589 ml/kg/h. Le somministrazioni sottocutanea ed endovenosa di Remodulin si sono dimostrate bioequivalenti allo stato stazionario nella dose di 10 ng/kg/min.
Metabolismo
Remodulin viene in gran parte metabolizzato a livello epatico. Il metabolismo di treprostinil coinvolge principalmente il CYP2C8 e in scarsa misura il CYP2C9. In base a dati in vitro relativi al citocromo P450 umano, Remodulin non inibisce CYP1A2, 2C9, 2C19, 2D6, 2E1 e 3A. Non sono stati condotti studi sulla possibile induzione degli isoenzimi del citocromo P450 umano da parte di Remodulin.
Non è stato osservato un singolo metabolita principale di treprostinil. Nell'urina sono stati rilevati 5 metaboliti, in quantità comprese tra il 10% e il 15% della dose somministrata, per un totale del 64%. 3 metaboliti sono prodotti dell'ossidazione della catena laterale del 3-idrossilottile, uno è un glucurono-coniugato e uno non è identificato. Solo il 3,7% della dose è stato rilevato immodificato nell'urina.
Eliminazione
L'emivita apparente di eliminazione di treprostinil in seguito alla somministrazione sottocutanea è di 1,3 ore rispetto a 45 minuti per l'infusione per via endovenosa.
In uno studio condotto in volontari sani utilizzando treprostinil 14C per un periodo di 224 ore, il 78,6% e il 13,4% della dose somministrata sono stati rilevati rispettivamente nell'urina e nelle feci.
Cinetica di gruppi di pazienti speciali
Disturbi della funzionalità epatica
L'esposizione plasmatica a treprostinil (area sottesa alla curva concentrazione plasmatica - tempo; AUC) aumenta del 260%-510% in caso di disturbi della funzionalità epatica da lievi a moderati, rispettivamente di classe A e B di Child-Pugh. La clearance plasmatica di treprostinil è risultata ridotta in misura fino all'80% nei soggetti che presentavano disturbi della funzionalità epatica da lievi a moderati rispetto ai soggetti sani (vedere «Posologia/impiego»).
Disfunzioni renali
Non sono stati condotti studi specifici. Un'analisi multivariata ha evidenziato che la clearance plasmatica di treprostinil è associata alla creatinina sierica.
Pazienti anziani
Nei pazienti anziani è stata osservata una leggera riduzione della clearance allo stato stazionario (circa 3%), che non giustifica tuttavia un aggiustamento della dose.
Bambini e adolescenti
Non sono stati condotti studi su bambini e adolescenti e pertanto l'impiego in questa popolazione non è raccomandato.
Impiego in caso di sovrappeso
I pazienti in sovrappeso (IMC >30 kg/m²) presentano una clearance inferiore di treprostinil.
Dati preclinici
Tossicità per somministrazione ripetuta
Negli studi condotti su ratti e conigli per un periodo di 26 settimane, treprostinil non è risultato tossico a dosi, rispettivamente, di 450 ng/kg/min e 200 ng/kg/min.
Mutagenicità
Gli studi di mutagenicità in vitro e in vivo non hanno mostrato effetti mutageni o clastogeni da parte del treprostinil.
Cancerogenicità
Non sono stati condotti studi a lungo termine per valutare il potenziale cancerogeno.
Tossicità per la riproduzione
Nei conigli è stato osservato un aumento del numero di variazioni scheletriche a dosi di 150 e 300 ng/kg/min e in presenza di tossicità materna. La somministrazione di dosi fino a 450 ng/kg/min non ha avuto effetti sulla capacità riproduttiva maschile e femminile nei ratti.
Altre indicazioni
Incompatibilità
Poiché non sono stati condotti studi di compatibilità, non si può somministrare questo medicamento in combinazione con altri medicamenti, eccetto con acqua per preparazioni iniettabili e soluzione acquosa di NaCl allo 0,9% (vedere « Indicazioni per la manipolazione»).
Stabilità
I flaconcini integri non deve essere utilizzato oltre la data indicata con «EXP» sul contenitore.
Stabilità dopo apertura
Una volta perforati, i flaconcini devono essere utilizzati entro 30 giorni.
Uso sottocutaneo
Il set per infusione (incluso il serbatoio della soluzione) deve essere sostituito ogni 48-72 ore.
Uso endovenoso
La soluzione diluita deve essere sostituita almeno ogni 48 ore.
Precauzioni particolari per la conservazione
Conservare a temperatura ambiente (15-25°C).
Tenere fuori dalla portata dei bambini.
Indicazioni per la manipolazione
Per la somministrazione endovenosa Remodulin deve essere diluito con acqua per preparazioni iniettabili o una soluzione acquosa di NaCl allo 0.9%. La compatibilità è dimostrata in un intervallo di concentrazione compreso tra 0,004 mg/ml e 0,13 mg/ml.
Per l'utilizzo della pompa vedere «Posologia/impiego».
Occorre tenere in considerazione che i pazienti con cateteri venosi centrali a lungo termine possono essere infettati da microrganismi Gram-negativi e Gram-positivi. Si raccomanda l'uso di un filtro in linea di 0,2 micrometri per ridurre al minimo il rischio di infezione sistemica causata da contaminazione durante la preparazione del medicamento.
Numero dell'omologazione
56120 (Swissmedic).
Titolare dell’omologazione
Gebro Pharma AG, 4410 Liestal.
Stato dell'informazione
Giugno 2020.
Guida alla posologia per la somministrazione sottocutanea
(Per la formula di calcolo vedere Posologia/impiego)




Guida alla posologia per la somministrazione endovenosa
(Per la formula di calcolo vedere Posologia/impiego)

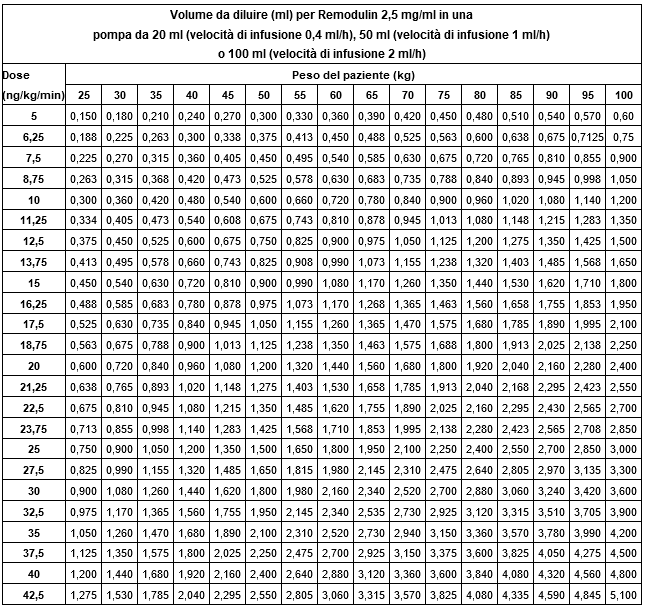


Composition
Principes actifs
Tréprostinil (sous forme sodique).
Excipients
Citrate de sodium dihydraté, acide chlorhydrique, métacrésol, hydroxyde de sodium, chlorure de sodium, eau pour préparations injectables.
Contenu total en sodium par mL
Dosage | 1.0 | 2.5 | 5.0 | 10.0 |
Contenu total en sodium (en mg) | 3.7 | 3.75 | 3.92 | 3.74 |
Forme pharmaceutique et quantité de principe actif par unité
Solution pour perfusion sous-cutanée ou intraveineuse:
Flacon perforable contenant 1 mg de tréprostinil/ml; 2,5 mg de tréprostinil/ml; 5 mg de tréprostinil/ml;10 mg de tréprostinil/ml.
Indications/Possibilités d’emploi
Traitement à long terme de l'hypertension pulmonaire primaire et de l'hypertension artérielle pulmonaire, associée à une maladie du tissu conjonctif, chez les patients à un stade clinique III ou IV de la NYHA (classification selon l'échelle de la New York Heart Association).
Posologie/Mode d’emploi
Le traitement par Remodulin sera instauré en milieu hospitalier sous surveillance médicale. Remodulin est administré en perfusion continue par voie sous-cutanée ou intraveineuse. En raison des risques associés aux cathéters veineux centraux à demeure, y compris de bactériémie et de septicémie grave, potentiellement mortelle, la perfusion sous-cutanée (sans dilution) est le mode d'administration à privilégier.
Instauration du traitement
Dose initiale
La perfusion sera débutée à raison de 1,25 ng/kg/min. Au cas où cette posologie ne serait pas tolérée par le patient, le taux de perfusion peut être ramené à 0,625 ng/kg/min.
Attention: ne jamais effectuer l'application intraveineuse non diluée.
Ajustement de la posologie/titration
Un ajustement posologique permanent vise à déterminer la dose permettant d'atténuer les symptômes de l'hypertension artérielle pulmonaire tout en maintenant le profil d'effets secondaires à un niveau acceptable. Dans les quatre premières semaines, le taux de perfusion doit être augmenté de 1,25 ng/kg/min par semaine, puis de 2,5 ng/kg/min par semaine.
Des effets indésirables dose-dépendants, tels que bouffées vasomotrices (flushing), céphalées, hypotension, nausées, vomissements et diarrhées, sont susceptibles d'imposer une réduction du taux de perfusion, mais peuvent éventuellement disparaître même sans ajustement de la dose. Si un effet indésirable s'aggrave et/ou devient intolérable, le taux de perfusion doit être réduit.
Eviter un arrêt soudain du traitement ou une forte réduction de la posologie (voir «Mises en garde et précautions»).
Après une interruption de quelques heures, la perfusion peut, dans la plupart des cas, être reprise avec les mêmes taux de perfusion. En cas d'interruption d'une durée plus longue, une nouvelle titration de la dose peut s'avérer nécessaire.
Dans les études d'efficacité de grande envergure, les taux de perfusion moyens après 12 mois était de 26 ng/kg/min, après 24 mois 36 ng/kg/min, après 36 mois 42 ng/kg/min.
Posologie usuelle
Perfusion continue par voie sous-cutanée
Le débit de perfusion sous-cutanée est calculé selon la formule suivante:
Débit de perfusion (ml/h):

* Facteur de conversion (0,00006): 60 min/h × 0,000001 mg/ng.
Perfusion continue par voie intraveineuse
Remodulin sous forme diluée est administré en perfusion intraveineuse continue par un cathéter veineux central à l'aide d'une pompe pour application intraveineuse.
Il peut également être administré transitoirement par une voie veineuse périphérique, de préférence dans une veine de gros calibre. L'utilisation d'une perfusion périphérique pendant plus de quelques heures peut augmenter le risque de thrombophlébite (voir «Effets indésirables»).
L'utilisation d'un filtre de 0,2 micromètre sur la ligne de perfusion est recommandée pour minimiser le risque d'infection systémique due à une contamination pendant la préparation du médicament.
Calcul de la concentration requise pour la solution diluée de Remodulin:
Concentration finale de la solution diluée de Remodulin (mg/ml):

Calcul de la quantité nécessaire de Remodulin devant être diluée par suffisamment de solvant (eau stérile ad inject. ou solution pour perfusions de NaCl à 0,9%):
Volume de Remodulin à appliquer (ml):

Mettre la quantité calculée de Remodulin dans le réservoir et le diluer avec suffisamment d'eau stérile ou de solution de NaCl à 0,9%.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
L'exposition systémique au tréprostinil (mesurée par l'aire sous la courbe des concentrations plasmatiques sur une période déterminée; AUC) était accrue de 260 à 510% chez les sujets présentant une insuffisance hépatique légère à modérée (stade A à B de la classification de Child-Pugh). Les troubles de la fonction hépatique légers à modérés réduisent la clairance plasmatique du tréprostinil de valeurs allant jusqu'à 80% par rapport aux sujets sains. Chez les patients atteints de troubles de la fonction hépatique, Remodulin ne doit donc être utilisé qu'avec précaution, le patient étant maintenu sous surveillance étroite pour dépister des effets secondaires dus à une concentration accrue de Remodulin (voir «Pharmacocinétique»).
La dose initiale de Remodulin doit être diminuée à 0,625 ng/kg/min. La dose doit ensuite être augmentée avec précaution.
Aucune expérience n'a été acquise concernant les patients atteints de troubles rénaux sévères.
Patients présentant des troubles de la fonction rénale
Aucune étude n'a été entreprise chez les patients souffrant de troubles de la fonction rénale. Le tréprostinil et ses métabolites étant principalement éliminés par voie urinaire, la prudence est recommandée lors du traitement de patients souffrant de troubles de la fonction rénale. Chez ces patients, une augmentation de la dose impose une plus grande prudence encore (voir « Pharmacocinétique, Posologie/Mode d'emploi»).
Application en cas de surcharge pondérale
Chez les patients présentant une surcharge pondérale (BMI >31 kg/m²), la clearance du tréprostinil se trouve réduite.
Patients âgés
On ne dispose pas de données suffisantes chez les patients âgés (≥65 ans). En raison de la fréquence accrue d'altérations des fonctions hépatique, rénale ou cardiaque, de maladies intercurrentes ou de traitements médicamenteux associés, la posologie doit être adaptée avec la plus grande prudence chez les sujets âgés.
Enfants et adolescents
On ne dispose que de peu de données chez les enfants et adolescents de moins de 16 ans pour pouvoir décider si administrer une dose d'adulte dans ce groupe de patients.
Remodulin est administré en perfusion continue par voie sous-cutanée ou intraveineuse. En raison des risques associés aux cathéters veineux centraux à demeure, y compris de bactériémie et de septicémie grave, potentiellement mortelle, la perfusion sous-cutanée (sans dilution) est le mode d'administration à privilégier.
Mode d'administration
Remodulin est administré en perfusion continue par voie sous-cutanée ou intraveineuse. En raison des risques associés aux cathéters veineux centraux à demeure, y compris de bactériémie et de septicémie grave, potentiellement mortelle, la perfusion sous-cutanée (sans dilution) est le mode d'administration à privilégier.
Dans la perfusion sous-cutanée, le cathéter est introduit sous la peau de l'abdomen ou de la cuisse, puis fixé. Lors de l'application sous-cutanée, le flacon perforable de Remodulin fait partie de la pompe. Il est impératif de remplacer, toutes les 48 à 72 heures, le matériel pour perfusion et le réservoir à médicament en question.
Dans la perfusion intraveineuse, un cathéter à demeure central est introduit. Ensuite, Remodulin est diffusé à un débit constant à l'aide d'une petite pompe attachée par une ceinture à la jambe, la cuisse ou l'abdomen. L'application intraveineuse fait appel à Remodulin dilué. La solution est transportée à travers un fin tuyau de polyvinyle vers l'aiguille.
Lors de la perfusion intraveineuse, il faut remplacer, toutes les 48 heures au moins, la solution diluée (en raison de la stabilité de Remodulin).
A l'hôpital, les patients sont instruits en détail, par le personnel soignant, sur l'emploi de la pompe, la mise en place du matériel pour perfusion, le remplacement du réservoir de Remodulin et l'administration de la perfusion continue. Respecter la manipulation hygiénique.
Le débit de Remodulin sera réajusté chaque semaine. En général, la posologie sera légèrement augmentée en fonction des symptômes de l'hypertension pulmonaire. A l'apparition d'effets secondaires intolérables, la posologie sera ajustée selon les instructions du médecin.
La manipulation correcte de la pompe pour perfusion est décrite dans son mode d'emploi.
Il importe d'utiliser Remodulin à une concentration correcte et le matériel pour perfusion approprié.
Un mauvais fonctionnement de la pompe ou une obturation du matériel pour perfusion risquent d'entraîner une aggravation des symptômes de l'hypertension artérielle pulmonaire. Au cas où le débit de Remodulin serait interrompu, suivre les instructions consignées dans le mode d'emploi de la pompe. Le patient doit toujours avoir une pompe de secours, un matériel pour perfusion et Remodulin en réserve.
La pompe à perfusion, utilisée pour l'application de Remodulin, doit répondre aux exigences suivantes:
- être de petite taille et de faible poids,
- permettre des ajustements de débit par paliers de 0,002 ml/h pour la perfusion s.c., ainsi que d'env. 0,05 ml/h pour la perfusion i.v. Pour les pompes à perfusion i.v., le taux de perfusion est typiquement compris entre 0,4 et 2 ml par heure,
- posséder une alarme pour les obstructions/les fins de perfusion, le déchargement de la pile, les défauts de programme ou les pannes du moteur,
- permettre une précision d'au moins ±6% ou plus par rapport au débit programmé,
- fonctionner par pression positive,
- le réservoir doit être en polychlorure de vinyle, en polypropylène ou en verre.
Remodulin est disponible à des concentrations de 1 mg/ml et 2,5 mg/ml, 5 mg/ml et 10 mg/ml.
Les tableaux comme aides de dosage figurent à la fin de l'information professionnelle.
Contre-indications
- Hypersensibilité à l'un des composants de Remodulin ou aux substances apparentées au tréprostinil.
- Hypertension artérielle pulmonaire liée à une maladie veino-occlusive,
- Insuffisance cardiaque congestive due à un dysfonctionnement sévère du ventricule gauche,
- Insuffisance hépatique sévère (Child-Pugh stade C),
- Ulcère gastro-intestinal en évolution, hémorragie intracrânienne, traumatisme récent ou autre état clinique susceptible d'entraîner une hémorragie,
- Anomalies valvulaires congénitales ou acquises avec retentissement cardiaque indépendamment de l'hypertension artérielle pulmonaire elle-même,
- Cardiopathie ischémique sévère ou angor instable; infarctus du myocarde survenu dans les six derniers mois; insuffisance cardiaque décompensée non surveillée médicalement; arythmies sévères; lésions cérébro-vasculaires (telles qu'accident ischémique transitoire ou accident vasculaire cérébral) survenues dans les trois derniers mois.
Mises en garde et précautions
Précautions générales
Remodulin ne doit être administré que par les médecins spécialisés, expérimentés dans le diagnostic et le traitement de l'hypertension artérielle pulmonaire. La décision d'instaurer un traitement par Remodulin devrait être prise en sachant que le traitement doit, selon toute vraisemblance, être pratiqué longtemps, éventuellement sur des années. A cet égard, la capacité du patient à appliquer Remodulin et à prendre soin du système à perfusion doit être prise en considération.
Remodulin s'administre uniquement par voie sous-cutanée ou sous forme diluée par voie intraveineuse.
Remodulin est un puissant vasodilatateur pulmonaire et systémique. Le traitement par Remodulin doit donc être instauré par un personnel disposant d'une formation adéquate, de même que d'un équipement requis pour une surveillance physiologique et un traitement d'urgence. Les ajustements posologiques ont été effectués dans des études cliniques, en raison des symptômes de l'hypertension artérielle pulmonaire et des effets secondaires de Remodulin.
Risques associés à l'administration intraveineuse de Remodulin
Des bactériémies et des septicémies associées aux cathéters veineux centraux et potentiellement mortelles ont été rapportées chez des patients ayant reçu Remodulin en perfusion intraveineuse. Ces risques sont imputables au mode d'administration. C'est pourquoi la perfusion sous-cutanée continue (sans dilution) est le mode d'administration à privilégier. La perfusion intraveineuse continue ne devra être envisagée que chez des patients stabilisés par perfusion sous-cutanée de tréprostinil mais qui ne tolèrent plus la voie sous-cutanée et si le niveau du risque encouru avec la voie intraveineuse centrale est jugé acceptable.
L'utilisation de techniques aseptiques est indispensable à la préparation et l'administration de Remodulin.
Risque d'hypotension systémique
Chez les patients présentant une pression artérielle systémique basse, le traitement par tréprostinil peut majorer le risque d'hypotension systémique. Il est recommandé de ne pas instaurer le traitement chez les patients dont la pression artérielle systolique est inférieure à 85 mmHg.
Lors de chaque modification de la dose, il est recommandé de surveiller la pression artérielle systémique et le rythme cardiaque afin d'arrêter la perfusion en cas d'hypotension ou de pression artérielle systolique inférieure ou égale à 85 mmHg.
Risques associés à une forte réduction de la dose ou un arrêt brusque du traitement
Comme avec tout vasodilatateur puissant, un arrêt brusque du traitement ou une forte réduction de la dose doivent être évités car ils risquent d'entraîner une aggravation de l'hypertension artérielle pulmonaire.
Risque d'hémorragies
Parce que ce groupe de patients était régulièrement traité par anticoagulants, des hémorragies sont fréquemment survenues. Les effets de Remodulin sur l'agrégation plaquettaire peuvent être à l'origine d'un accroissement du risque hémorragique comme cela a été observé par l'augmentation de l'incidence des événements à type d'épistaxis et d'hémorragies gastro-intestinales (incluant hémorragies rectales, gingivales et méléna) dans les essais cliniques contrôlés. Des hémoptysies, des hématémèses et des hématuries ont également été rapportées, mais avec une fréquence inférieure ou égale à celle dans le groupe traité par placebo.
Administration concomitante d'autres médicaments
L'administration concomitante d'un inhibiteur enzymatique du cytochrome P450 (CYP2C8) (p.ex. gemfibrozil) peut augmenter l'exposition (à la fois la Cmax et l'AUC) au tréprostinil. Une exposition accrue risque d'entraîner une augmentation des effets indésirables associés à l'administration du tréprostinil. Dans ce cas, il convient d'envisager une réduction de la dose de tréprostinil (voir «Interactions»).
L'administration concomitante d'un inducteur de l'enzyme CYP2C8 (p.ex. rifampicine) peut diminuer l'exposition au tréprostinil. Une diminution de l'exposition risque d'entraîner une réduction de l'efficacité clinique. Dans ce cas, il convient d'envisager une augmentation de la dose de tréprostinil (voir «Interactions»).
Le rapport bénéfice/risque de Remodulin n'a pas été étudié dans les hypertensions artérielles pulmonaires associées à un shunt cardiaque gauche-droit, une hypertension portale ou une infection par le VIH.
Remodulin contient au max. 75 mg (dosage de 5 mg/ml) de sodium par flacon de 20 ml, ce qui équivaut à 3,75% de l'apport alimentaire quotidien maximal recommandé par l'OMS de 2 g de sodium par adulte. Ceci doit être pris en considération chez les patients suivant un régime hyposodé strict.
Interactions
Au cours des études cliniques, Remodulin a été utilisé en même temps que les médicaments suivants: anticoagulants, diurétiques, glucosides cardiotoniques, antagonistes du calcium, analgésiques, antipyrétiques, antiphlogistiques non stéroïdiens, corticostéroïdes et autres.
L'administration concomitante de Remodulin avec les diurétiques, les agents d'antihypertenseurs ou d'autres vasodilatateurs majore le risque d'hypotension systémique.
Lorsque d'autres inhibiteurs de l'agrégation plaquettaire ou anticoagulants sont administrés en même temps, Remodulin est susceptible d'augmenter le risque d'hémorragies.
Interactions pharmacocinétiques
Données in vivo
Acétaminophène: Des études d'interactions ont été effectuées en utilisant l'acétaminophène (4 g/jour) chez des sujets sains. L'acétaminophène n'a exercé aucune action clinique significative sur la pharmacocinétique du tréprostinil. Aucune interaction avec le phenprocoumone n'est attendue sur la base des études in vitro qui ont été menées sur les microsomes hépatiques.
Furosémide: Chez les patients ayant reçu du furosémide, la clairance plasmatique a été réduite. Il est possible qu'il existe une interaction entre le tréprostinil et le furosémide, les deux substances étant soumises à une glucuronoconjugaison, pendant la métabolisation au groupe carboxylate.
Bosentan: Dans une étude de pharmacocinétique humaine conduite avec le bosentan (250 mg/jour) et le tréprostinil diolamine (dose orale de 2 mg/jour), aucune interaction pharmacocinétique n'a été observée entre le tréprostinil et le bosentan. Il n'a pas établi si la sécurité et l'efficacité de Remodulin, administré par voie parentérale (sous-cutanée ou intraveineuse), sont modifiées sous l'effet de bosentan.
Sildénafil: Dans une étude de pharmacocinétique humaine conduite avec le sildénafil (60 mg/jour) et le tréprostinil diolamine (dose orale de 2 mg/jour), aucune interaction pharmacocinétique n'a été observée entre le tréprostinil et le sildénafil. Il n'a pas établi si la sécurité et l'efficacité de Remodulin, administré par voie parentérale (sous-cutanée ou intraveineuse), sont modifiées sous l'effet de sildénafil.
Interactions pharmacodynamiques
Inhibiteurs de l'agrégation plaquettaire, y compris anti-inflammatoires non stéroïdiens (AINS) et anticoagulants
Le tréprostinil peut exercer un effet inhibiteur sur la fonction plaquettaire. L'administration concomitante de Remodulin avec des agents anti-agrégants plaquettaires, y compris les AINS, les agents dits «donneurs de monoxyde d'azote» et les anticoagulants, peut majorer le risque hémorragique. Les patients sous anticoagulants devront être régulièrement contrôlés selon les modalités conventionnelles de la pratique médicale en matière de surveillance. L'utilisation concomitante d'antiagrégants plaquettaires supplémentaires doit être évitée chez les patients traités par anticoagulants. Une perfusion continue sous-cutanée de tréprostinil n'a pas eu d'impact sur les propriétés pharmacodynamiques ou pharmacocinétiques d'une dose unique de warfarine (25 mg).
A ce jour, aucune donnée clinique ne confirme l'augmentation du risque hémorragique en cas d'administration concomitante de tréprostinil et de médicaments dits «donneurs de monoxyde d'azote».
Inducteurs enzymatiques
Rifampicine: des études de pharmacocinétique humaine conduites avec le tréprostinil diolamine administré par voie orale ont montré que la co-administration de la rifampicine, inducteur enzymatique du CYP2C8, réduit l'exposition au tréprostinil (d'environ 20- 30 %). Il n'a pas établi si la sécurité et l'efficacité de Remodulin, administré par voie parentérale (sous-cutanée ou intraveineuse), sont modifiées sous l'effet de la rifampicine. Si la rifampicine est ajoutée ou retirée de la médication d'un patient après la période de titration, il convient d'envisager un ajustement de la dose de tréprostinil.
Les inducteurs du CYP2C8 (p.ex. phénytoïne, carbamazépine, phénobarbital et millepertuis) peuvent réduire l'exposition au tréprostinil. Si un inducteur du CYP2C8 est ajouté ou retiré de la médication d'un patient après la période de titration, il convient d'envisager un ajustement de la dose de tréprostinil.
Inhibiteurs enzymatiques
Gemfibrozil: des études de pharmacocinétique humaine conduites avec le tréprostinil diolamine administré par voie orale ont montré que la co-administration du gemfibrozil, inhibiteur enzymatique du cytochrome P450 (CYP2C8), double l'exposition au tréprostinil (à la fois Cmax et AUC). Il n'a pas établi si la sécurité et l'efficacité de Remodulin, administré par voie parentérale (sous-cutanée ou intraveineuse), sont modifiées sous l'effet des inhibiteurs du CYP2C8. Si un inhibiteur du CYP2C8 (p.ex. gemfibrozil, triméthoprime et déférasirox) est ajouté ou retiré de la médication d'un patient après la période de titration, il convient d'envisager un ajustement de la dose de tréprostinil.
Grossesse/Allaitement
Grossesse
Chez le lapin, un nombre accru d'anomalies squelettiques a été retrouvé au niveau foetal lors d'une toxicité maternelle (à des doses de 150 et 300 ng/kg/min). A part cela, aucun effet indésirable n'a été établi dans des études portant sur la toxicité de reproduction.
Il n'y a pas d'études disponibles chez la femme enceinte. Remodulin ne doit donc être utilisé pendant la grossesse qu'en cas de nécessité absolue.
Allaitement
On ignore si le tréprostinil diffuse dans le lait maternel et s'il est absorbé au niveau systémique par l'enfant. Par conséquent, Remodulin ne doit pas être utilisé pendant la période d'allaitement.
Effet sur l’aptitude à la conduite et l’utilisation de machines
L'initiation du traitement ou les phases d'ajustement de la posologie peuvent être accompagnées d'effets indésirables tels qu'une hypotension systémique symptomatique ou des sensations de vertige pouvant altérer l'aptitude et l'assurance quant à la conduite de véhicules et l'utilisation de machines. En raison d'effets indésirables éventuels, la prudence est de rigueur lorsqu'il s'agit de conduire des véhicules ou d'utiliser des machines.
Effets indésirables
Une interprétation des effets indésirables est compliquée par une similitude entre les divers symptômes cliniques individuels de l'hypertension artérielle pulmonaire (p.ex. vertiges, évanouissement). Les effets indésirables pouvant probablement être attribués à la maladie de fond comprennent: détresse respiratoire, fatigue, douleurs thoraciques, infarctus du ventricule droit et pâleur.
Certains effets indésirables peuvent sans doute être imputés à Remodulin. Les plus fréquents d'entre eux lors de l'administration sous-cutanée sont des réactions au site de perfusion (83%) et des douleurs au site de perfusion (85%); elles sont cependant tolérées par la majorité des patients. Tous les événements tels qu'érythèmes, indurations, éruptions cutanées, etc., à l'exception des douleurs ou hémorragies/bleus, sont attribués aux réactions au site de perfusion.
Les effets indésirables sont rangés par classe de système d'organes de la classification MedDRA et par fréquence selon la convention suivante:
«très fréquents» (≥1/10),
«fréquents» (≥1/100 à <1/10),
«occasionnels» (≥1/1000 à <1/100),
«rares» (≥1/10 000 à <1/1000),
«très rares» (<1/10 000),
«Fréquence inconnue» (ne peut être estimée sur la base des données disponibles).
Infections et infestations
Fréquence inconnue: infection systémique liée au cathéter veineux central, septicémie, bactériémie**, infection au site de perfusion, formation d'abcès au site de perfusion sous-cutanée, cellulite.
Affections hématologiques et du système lymphatique
Fréquence inconnue: thrombocytopénie.
Affections du système nerveux
Très fréquents: céphalées (30%).
Fréquents: vertiges.
Affections cardiaques
Fréquence inconnue: insuffisance cardiaque à haut débit.
Affections vasculaires
Très fréquents: vasodilatations/bouffées vasomotrices (flushing) (11%).
Fréquents: hypotension, accident hémorragique§.
Fréquence inconnue: thrombophlébite*.
Affections gastro-intestinales
Très fréquents: diarrhées (22%), nausées (19%).
Fréquents: vomissements.
Affections de la peau et du tissu sous-cutané
Très fréquents: éruption cutanée (12%).
Fréquents: prurit.
Fréquence inconnue: éruptions cutanées généralisées de nature maculaire ou papuleuse.
Affections musculosquelettiques et du tissu conjonctif
Très fréquents: douleurs de la mâchoire (13%).
Fréquents: myalgie, arthralgie, douleurs aux extrémités.
Fréquence inconnue: douleurs osseuses.
Troubles généraux et anomalies au site d'administration
Très fréquents: douleurs au site de perfusion (85%), réaction au site de perfusion (83%), saignement ou hématome (21%).
Fréquents: œdèmes.
* Des cas de thrombophlébite associée à la perfusion intraveineuse périphérique ont été rapportés.
** Des cas menaçant le pronostic vital et mortels ont été rapportés.
§ Voir la rubrique «Description d'effets indésirables sélectionnés»
Description d'effets indésirables sélectionnés
Incidents hémorragiques
Les hémorragies ont été fréquentes en raison du nombre important de patients traités par anticoagulants. Les effets de Remodulin sur l'agrégation plaquettaire peuvent être à l'origine d'un accroissement du risque hémorragique comme cela a été observé par l'augmentation de l'incidence des événements à type d'épistaxis et de saignements digestifs (incluant hémorragies gastro-intestinales, hémorragie rectale, palatine et méléna) dans des essais cliniques contrôlés. Des hémoptysies, des hématémèses et des hématuries ont également été rapportées, mais avec une fréquence inférieure ou égale à celle dans le groupe traité par placebo.
Événements indésirables liés au mode d'administration par voie intraveineuse
L'administration de Remodulin en perfusion intraveineuse par des cathéters veineux centraux a été étudiée dans un nombre limité d'études cliniques. Dans ces études, le profil général d'événements indésirables est – de manière prévisible – similaire à celui lors de l'administration sous-cutanée de Remodulin compte tenu de la bioéquivalence des modes d'administration intraveineux et sous-cutané, qui a pu être mise en évidence. Le port à long terme d'un cathéter veineux central comporte toutefois certains risques associés à ce mode d'administration comprenant douleurs au site d'insertion du cathéter, infections locales, septicémie, formation de thrombus avec la maladie occlusive des vaisseaux qui en résulte ainsi que dysfonctionnements du système d'administration entraînant l'administration accidentelle d'un bolus ou une réduction involontaire de la dose. Du fait du surdosage ou du sous-dosage de Remodulin, l'apparition des symptômes correspondants est alors possible.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
Durant les études cliniques, les signes et symptômes d'un surdosage de Remodulin étaient similaires aux effets pharmacologiques susceptibles de limiter l'augmentation des doses, tels que bouffées vasomotrices (flushing), céphalées, hypotension, nausées, vomissements et diarrhées. La plupart de ces effets étaient autolimitatifs et ont disparu après une réduction de la dose ou un arrêt du traitement. Dans des cas isolés, une décompensation de l'hypertension artérielle pulmonaire a également été observée.
Propriétés/Effets
Code ATC
B01AC
Remodulin est une prostacycline synthétique.
Mécanisme d'action
Les effets pharmacologiques principaux du tréprostinil résident dans deux actions: vasodilatation directe des lits vasculaires artériels, pulmonaires et systémiques, et inhibition de l'agrégation plaquettaire. Chez l'animal, les effets vasodilatateurs réduisent la postcharge des ventricules droit et gauche tout en augmentant le débit cardiaque et le volume d'éjection systolique. Chez l'animal, l'effet du tréprostinil sur le rythme cardiaque dépend de la dose administrée. Il n'a pas été observé d'effet notable sur la conduction cardiaque.
Pharmacodynamique
L'ensemble des résultats de deux études fondamentales randomisées, effectuées en double insu et contrôlées par placebo, a révélé qu'après un traitement de deux semaines par Remodulin, associé au traitement classique, l'amélioration moyenne des paramètres hémodynamiques suivants, comparée aux valeurs de référence, était plus marquée que sous traitement par placebo, associé au traitement classique: réduction de la pression artérielle pulmonaire maximale (PAPm), de la résistance artérielle pulmonaire (RAP), de l'indice de la résistance vasculaire pulmonaire (IRVP) et de l'indice de la résistance vasculaire systémique (IRVS) ainsi qu'augmentation de la capacité inspiratoire et de la saturation du sang veineux en oxygène.
Efficacité clinique
La mobilité des patients a été mesurée, pendant six minutes, à l'aide d'un test de marche; chez les patients ayant reçu, conjointement avec un traitement classique, une perfusion de Remodulin, administrée sur 12 mois par voie sous-cutanée (n= 232), cette mobilité s'est avérée meilleure (valeur médiane de 10 vs 0 m) que chez les sujets maintenus sous placebo, associé au traitement classique (n= 236) (p= 0,0064). Cette amélioration a déjà été perceptible après 6 semaines de traitement. La meilleure mobilité a été liée à un soulagement statistiquement significatif de la dyspnée et de la fatigue, évalué selon l'échelle dyspnée – fatigue de Borg, de même que d'une hypertension artérielle pulmonaire.
Pharmacocinétique
Absorption
Lorsque Remodulin est perfusé par voie sous-cutanée, les taux plasmatiques en état d'équilibre sont normalement atteints entre 15 et 18 heures après le début du traitement et s'avèrent, lors d'un taux de perfusion de 2,5 jusqu'à 125 ng/kg/min, proportionnels à la dose administrée. En perfusion sous-cutanée, Remodulin est intégralement absorbé.
Distribution
Chez les sujets sains, les concentrations plasmatiques ont atteint après administration sous-cutanée leur maximum à 1 heure la nuit et à 10 heures du matin, et leur minimum à 7 heures du matin et à 16 heures. Les valeurs maximales ont été de quelque 20 à 30% supérieures aux valeurs minimales. Un ajustement de la dose en raison des fluctuations journalières n'est pas jugé nécessaire.
Le volume de distribution moyen est de l'ordre de 1,1 l/kg, la clearance plasmatique de 589 ml/kg/h.
Les administrations sous-cutanée et intraveineuse de Remodulin se sont montrées bioéquivalentes à l'état d'équilibre avec une dose de 10 ng/kg/min.
Métabolisme
Remodulin est métabolisé, en grande partie, au niveau hépatique. Le tréprostinil est essentiellement métabolisé par le CYP2C8 et, dans une moindre mesure, par le CYP2C9. En raison de données obtenues in vitro avec les isoenzymes P450 humaines, Remodulin n'inhibe pas les CYP1A2, 2C9, 2C19, 2D6, 2E1 et 3A. La question de savoir si Remodulin induit les isoenzymes P450 n'a pas encore fait l'objet de recherches chez l'homme.
Aucun métabolite principal individuel du tréprostinil n'a été observé. Cinq métabolites ont été identifiés dans les urines, en quantité allant de 10 jusqu'à 15% de la dose administrée, 64% au total. Trois de ces métabolites sont des produits d'oxydation de la chaîne 3-hydroxylactylique, un d'entre eux est un glucuronoconjugué et l'un n'a pas été identifié. Seuls 3,7% du principe actif ont été retrouvés sous forme inchangée dans les urines.
Élimination
En application sous-cutanée, le tréprostinil présente une demi-vie d'élimination apparente de 1,3 h comparée à 45 minutes lors de l'administration intraveineuse.
Dans une étude effectuée chez les sujets sains ayant reçu du tréprostinil marqué au C14, 78,6% de la dose administrée ont été retrouvés, sur une période de 224 heures, dans les urines et 13,4% dans les fèces.
Cinétique pour certains groupes de patients
Patients présentant des troubles de la fonction hépatique
L'exposition systémique au tréprostinil (mesurée par l'aire sous la courbe des concentrations plasmatiques sur une période déterminée; AUC) a augmenté de 260 à 510% chez les sujets présentant une insuffisance hépatique légère à modérée (stade A à B de la classification de Child-Pugh). La clairance plasmatique chez les patients présentant une insuffisance hépatique légère à modérée était diminuée de jusqu'à 80% par rapport à celle observée chez des adultes sains (voir «Posologie/Mode d'emploi»).
Patients présentant des troubles de la fonction rénale
On ne dispose pas d'études particulières. Une analyse de variantes multiples a révélé que la clairance plasmatique du tréprostinil est associée à la créatinine sérique.
Patients âgés
Une réduction minime de la clearance en état d'équilibre (3%) a été observée chez les malades âgés; elle ne justifie cependant guère un ajustement de la dose.
Enfants et adolescents
Aucune étude n'ayant été effectuée chez l'enfant et l'adolescent, l'emploi de Remodulin n'est pas recommandé.
Application en cas de surcharge pondérale
Chez les patients présentant une surcharge pondérale (BMI >31 kg/m²), la clearance du tréprostinil se trouve réduite.
Données précliniques
Toxicité en cas d'administration répétée
Dans les études réalisées pendant 26 semaines sur le rat et le lapin, le tréprostinil, administré à une posologie de 450 ng/kg/min ou 200 ng/kg/min, s'est avéré non toxique.
Mutagénicité
Les tests de mutagénicité in vitro et in vivo n'ont révélé aucun effet mutagène ni clastogène du tréprostinil.
Carcinogénicité
Aucune étude à long terme permettant d'évaluer l'effet carcinogène n'a été effectuée.
Toxicité sur la reproduction
A des doses allant jusqu'à 900 ng/kg/min chez le rat et jusqu'à 50 ng/kg/min chez le lapin, il n'a été observé aucun effet toxique sur la reproduction. Chez le lapin, un nombre accru d'anomalies squelettiques a été retrouvé à des doses de 150 et 300 ng/kg/min et lors d'une toxicité maternelle. Les doses allant jusqu'à 450 ng/kg/min n'ont pas modifié la capacité de reproduction chez le rat, mâle ou femelle.
Remarques particulières
Incompatibilités
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé à d'autres médicaments, sauf avec eau pour préparations injectables et solution aqueuse de NaCl à 0,9% (voir «Remarques concernant la manipulation»).
Stabilité
Les flacons perforable non entamés ne doivent pas être utilisés au-delà de la date indiquée sur l'emballage par la mention «EXP».
Stabilité après ouverture
Les flacons une fois percés peuvent s'utiliser pendant 30 jours au maximum.
Application sous-cutanée
Remplacer, toutes les 48 à 72 heures, le matériel pour perfusion (le réservoir à solution compris).
Application intraveineuse
Il est impératif de remplacer toutes les 48 heures la solution diluée.
Remarques particulières concernant le stockage
Conserver à température ambiante (15-25°C).
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Pour l'administration intraveineuse, Remodulin doit être dilué avec de l'eau pour préparations injectables ou une solution aqueuse de NaCl à 0,9%. La compatibilité est étayée dans une fourchette de concentrations de 0,004 mg/ml à 0,13 mg/ml.
Pour l'utilisation de la pompe, voir «Posologie/Mode d'emploi».
Il faut prendre en compte le fait que les patients portant un cathéter veineux central sur une longue durée présentent un risque de contamination par les germes Gram négatif et Gram positif. Il est recommandé d'utiliser un filtre de 0,2 micron sur la ligne de perfusion afin de minimiser le risque d'infection systémique qui résulterait d'une contamination au moment de la préparation du médicament.
Numéro d’autorisation
56120 (Swissmedic).
Titulaire de l’autorisation
Gebro Pharma AG, 4410 Liestal.
Mise à jour de l’information
Juin 2020.
Aide de dosage pour application s.c.
(formule calcul voir Posologie/Mode d’emploi)




Aide de dosage pour application i.v.
(formule calcul voir Posologie/Mode d’emploi)




Отзывов (0)
Вы смотрели



Бесплатная консультация опытного специалиста
Опишите симптомы или нужный препарат – мы поможем подобрать его дозировку или аналог, оформим заказ с доставкой на дом или просто проконсультируем.
Нас 14 специалистов и 0 ботов. Мы всегда будем с вами на связи и сможем связаться в любое время.
Хит продаж


ГерпоТерм ручка от герпеса
Код продукта: 7798882Herpotherm® — нагревательная ручка Герпес не только некрасив, но и может быть очень болезненным. Со..
8364.60 RUB








Hydroclean Plus Wundkissen 5.5см Rund 10 штук
Код продукта: 7807653..
13849.21 RUB

Бургерштейн Витамин Д3 800 МЕ спрей 20 мл
Код продукта: 7767304Бургерштейн Витамин Д3 - это пищевая добавка, которая компенсирует недостаточную выработку орган..
4816.69 RUB

Бургерштейн Доломит 240 таблеток
Код продукта: 1587415Бюргерштейн Доломит - это пищевая добавка с оптимальным соотношением кальция и магния. Эти два ми..
6844.00 RUB


Бускопан 10 мг 40 драже
Код продукта: 5304383Что такое Бускопан и когда он используется?Бускопан оказывает спазмолитическое и, следовательно, об..
7668.17 RUB


ВитроКап Н 90 касул
Код продукта: 7803065ВитроКап Н — это пищевая добавка, содержащая уникальную смесь необходимых витаминов и минералов дл..
13074.93 RUB


Остенил вязкоупругий имплантат для внутрисуставных инъекций стерильный 20 мг/2 мл, шприц одноразовый 2 мл
Код продукта: 2055241СоставОстенил, Остенил Мини1 мл изотонического раствора (рН 7,3) содержит 10,0 мг гиалуроновой..
11024.97 RUB




Сидрога детский чай от бронхита 20 пакетиков
Код продукта: 2025240ФитотерапияЧто Sidroga детей Bronchialtee и когда он применяется?Sidroga детей Bronchialtee содержит..
1979.61 RUB


Тавегил 1 мг 20 таблеток
Код продукта: 388056Что такое Тавегил и когда он используется?Тавегил содержит активный ингредиент клемастин, антигис..
5575.03 RUB