



Алпроликс сухое вещество 2000 Ie Cum Solv флакон
ALPROLIX Trockensub 2000 IE cum Solv
-
510697.59 RUB

- Наличие: Нет в наличии
- Модель: 6850018
- ATC-код B02BD04
- EAN 7680660390043
Состав:
Наведите телефон на qr-код

Описание
 Deutsch
Deutsch French
French Italian
ItalianZusammensetzung
Wirkstoffe
Eftrenonacog alfa (humanen Gerinnungsfaktor IX (rDNA))
Hilfsstoffe
Pulver: Saccharose, Histidin, Mannitol, Polysorbat 20, Natriumhydroxid (zur pH-Einstellung), Salzsäure (zur pH-Einstellung)
Lösungsmittel: Natriumchloridlösung, Wasser für Injektionszwecke
Natrium je Durchstechflasche: 0,3 mmol (6,4 mg)
Darreichungsform und Wirkstoffmenge pro Einheit
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung (i.v.).
1 Durchstechflasche Pulver ALPROLIX enthält Eftrenonacog alfa in den folgenden Konzentrationen:
250 I.E. Eftrenonacog alfa (50 I.E./ml nach Rekonstitution mit 5 ml Lösungsmittel (Natriumchloridlösung))
500 I.E. Eftrenonacog alfa (100 I.E./ml nach Rekonstitution mit 5 ml Lösungsmittel (Natriumchloridlösung))
1000 I.E. Eftrenonacog alfa (200 I.E./ml nach Rekonstitution mit 5 ml Lösungsmittel (Natriumchloridlösung))
2000 I.E. Eftrenonacog alfa (400 I.E./ml nach Rekonstitution mit 5 ml Lösungsmittel (Natriumchloridlösung))
3000 I.E. Eftrenonacog alfa (600 I.E./ml nach Rekonstitution mit 5 ml Lösungsmittel (Natriumchloridlösung))
Indikationen/Anwendungsmöglichkeiten
ALPROLIX ist indiziert für die Behandlung und Prophylaxe von Blutungen bei Patienten mit Hämophilie B (kongenitaler Faktor-IX-Mangel).
Dosierung/Anwendung
Die Behandlung sollte unter Aufsicht eines in der Hämophiliebehandlung erfahrenen Arztes eingeleitet werden.
Therapieüberwachung
Während der Therapie wird eine geeignete Bestimmung der Faktor-IX-Spiegel empfohlen, um die anzuwendende Dosierung und Häufigkeit von wiederholten Injektionen zu steuern. Das Ansprechen einzelner Patienten auf Faktor IX kann variieren, was sich in unterschiedlichen Halbwertszeiten und verschiedenen Recoveries äussert. Bei der auf dem Körpergewicht basierenden Dosierung kann eine Anpassung für unter- oder übergewichtige Patienten erforderlich sein. Insbesondere bei grösseren chirurgischen Eingriffen ist eine genaue Überwachung der Substitutionstherapie mittels Gerinnungsanalyse (Faktor-IX-Aktivität im Plasma) unerlässlich.
Bei Verwendung eines einstufigen In-vitro-Gerinnungstests auf Basis der Thromboplastinzeit (aPTT) zur Bestimmung der Faktor-IX-Aktivität in Blutproben des Patienten können die Ergebnisse der Faktor-IX-Aktivität sowohl von der Art des verwendeten aPTT-Reagenz als auch vom verwendeten Referenzstandard erheblich beeinflusst werden. Dies ist besonders dann zu beachten, wenn das Labor und/oder die im Test verwendeten Reagenzien gewechselt werden.
Messungen mit einem Einstufen-Gerinnungstest, in dem ein kaolinbasiertes aPTT-Reagenz verwendet wird, führen wahrscheinlich zu einer Unterbewertung des Aktivitätsspiegels.
Dosierung
Dosierung und Dauer der Substitutionstherapie richten sich nach dem Schweregrad des Faktor-IX-Mangels, der Lokalisation und dem Ausmass der Blutung sowie nach dem klinischen Zustand des Patienten.
Die Anzahl der anzuwendenden Einheiten des Faktors IX wird in Internationalen Einheiten (I.E.) auf Basis des derzeitigen WHO-Standards für Faktor-IX-Präparate angegeben. Die Faktor-IX-Aktivität im Plasma wird entweder als Prozentsatz (bezogen auf normales menschliches Plasma) oder in Internationalen Einheiten (bezogen auf einen internationalen Standard für Faktor IX im Plasma) dargestellt.
Eine Internationale Einheit (I.E.) des rekombinanten Faktors IX Fc entspricht der Menge Faktor IX in einem ml normalem menschlichem Plasma.
Bedarfsbehandlung
Die Berechnung der benötigten Dosis des rekombinanten Faktors IX Fc basiert auf dem empirischen Befund, dass 1 Internationale Einheit (I.E.) Faktor IX pro kg Körpergewicht die Faktor-IX-Aktivität im Plasma um 1% der normalen Aktivität (I.E./dl) erhöht. Die erforderliche Dosis wird anhand folgender Formel berechnet:
Erforderliche Einheiten = Körpergewicht (kg) x gewünschter Faktor-IX-Anstieg (%) (I.E./dl) x Kehrwert der ermittelten Recovery (I.E./kg pro I.E./dl).
Die erforderliche Dosis und die Häufigkeit der Anwendung sollten sich stets an der klinischen Wirksamkeit im Einzelfall orientieren. Falls zur Kontrolle der Blutung eine wiederholte Gabe erforderlich ist, sollte die längere Halbwertszeit von ALPROLIX berücksichtigt werden (siehe „Pharmakokinetik”). Es wird nicht erwartet, dass sich die Zeit bis zur maximalen Aktivität verzögert.
Bei den nachfolgend beschriebenen Blutungsereignissen sollte die Faktor-IX-Aktivität im entsprechenden Zeitraum nicht unter die angegebene Plasmaaktivität (in % der Norm oder in I.E./dl) abfallen. Die Angaben in Tabelle 1 können als Richtwerte für die Dosierung bei Blutungsepisoden und chirurgischen Eingriffen verwendet werden.
Tabelle 1: Richtlinien für die Dosierung von ALPROLIX zur Behandlung von Blutungsepisoden und bei chirurgischen Eingriffen
Schweregrad der Blutung/Art des chirurgischen Eingriffs | Erforderlicher Faktor-IX-Spiegel (%) (I.E./dl) | Häufigkeit der Anwendung (Stunden)/Therapiedauer (Tage) |
Blutung | ||
Hämarthrose im Frühstadium, Muskelblutung oder orale Blutung | 20-40 | Injektion alle 48 Stunden wiederholen, bis die Blutungsepisode – angezeigt durch Schmerzen – abgeklungen ist oder eine Heilung erreicht wurde.1 |
Umfangreichere Hämarthrose, Muskelblutung oder Hämatom | 30-60 | Injektion alle 24 bis 48 Stunden wiederholen, bis Schmerzen und akute Beeinträchtigungen abgeklungen sind.1 |
Lebensbedrohende Blutungen | 60-100 | Injektion alle 8 bis 24 Stunden wiederholen, bis der Patient ausser Lebensgefahr ist. |
Chirurgische Eingriffe | ||
Kleinere Eingriffe einschliesslich Zahnextraktion | 30-60 | Wiederholung der Injektion alle 24 bis 48 Stunden soweit erforderlich, bis eine Heilung erreicht wurde. |
Grössere chirurgische Eingriffe | 80-100 (prä- und postoperativ) | Injektion alle 8 bis 24 Stunden soweit erforderlich bis zur ausreichenden Wundheilung wiederholen, danach Therapie über mindestens 7 Tage fortsetzen, um eine Faktor-IX-Aktivität von 30% bis 60% (I.E./dl) aufrechtzuerhalten. |
1 Siehe Tabelle 3 im Abschnitt „Pharmakokinetik”.
Prophylaxe
Für die Langzeitprophylaxe von Blutungen werden die folgenden initialen Dosierungsschemata empfohlen:
• 50 I.E./kg einmal wöchentlich, Dosisanpassung an das individuelle Ansprechen des Patienten oder
• 100 I.E./kg alle 10 Tage, Anpassung des Intervalls an das individuelle Ansprechen des Patienten. Einige Patienten, die mit einem Behandlungsregime mit 10-tägigem Intervall gut eingestellt sind, können möglicherweise in einem Intervall von 14 oder mehr Tagen behandelt werden.
Die höchste empfohlene Dosis für die Prophylaxe ist 100 I.E./kg.
Spezielle Dosierungsanweisungen
Ältere Patienten
Die Erfahrungen bei Patienten im Alter von ≥65 Jahren sind begrenzt.
Kinder und Jugendliche
Bei Kindern unter 12 Jahren können höhere Dosen oder kürzere Dosierungsintervalle erforderlich sein. Die empfohlene Initialdosis ist 50-60 I.E./kg alle 7 Tage. Für Jugendliche ab 12 Jahren gelten die gleichen Dosierungsempfehlungen wie für Erwachsene (siehe «Eigenschaften/Wirkungen» und «Pharmakokinetik». Die höchste empfohlene Dosis für die Prophylaxe ist 100 I.E./kg.
Es gibt für Kinder im Alter unter 6 Jahren nur in begrenztem Umfang Daten zur Behandlung bei chirurgischen Eingriffen.
Art der Anwendung
Intravenöse Anwendung.
ALPROLIX sollte über mehrere Minuten intravenös injiziert werden. Die Verabreichungsgeschwindigkeit sollte sich nach dem Befinden des Patienten richten und 10 ml/min nicht überschreiten.
Die Anwendung als kontinuierliche Infusion ist nicht zugelassen und wird nicht empfohlen (siehe auch «Warnhinweise und Vorsichtsmassnahmen»).
Für Hinweise zur Rekonstitution des Arzneimittels vor der Anwendung, siehe «Hinweise für die Handhabung».
Dokumentation der Chargennummer
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Kontraindikationen
Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe.
Warnhinweise und Vorsichtsmassnahmen
Überempfindlichkeit
Unter der Therapie mit ALPROLIX ist von allergischen Überempfindlichkeitsreaktionen berichtet worden. Die Patienten sind anzuweisen, die Anwendung des Arzneimittels sofort abzubrechen und sich an ihren Arzt zu wenden, falls Symptome einer Überempfindlichkeitsreaktion auftreten. Die Patienten sollten über die ersten Anzeichen von Überempfindlichkeitsreaktionen einschliesslich Nesselsucht, generalisierter Urtikaria, Engegefühl in der Brust, Giemen, Hypotonie und Anaphylaxie informiert werden.
Im Fall eines anaphylaktischen Schocks sollte die medizinische Standardtherapie zur Schockbehandlung eingeleitet werden.
Inhibitoren
Nach wiederholter Behandlung mit Faktor-IX-Präparaten sollten die Patienten auf die Entwicklung von neutralisierenden Antikörpern (Inhibitoren) überwacht werden. Diese sollten mittels geeigneter biologischer Tests in Bethesda-Einheiten (B.E.) quantifiziert werden.
Berichte in der Fachliteratur zeigen eine Korrelation zwischen dem Auftreten eines Faktor-IX-Inhibitors und allergischen Reaktionen. Daher sollten Patienten, bei denen allergische Reaktionen auftreten, auf Inhibitoren getestet werden. Weiterhin sollte bedacht werden, dass Patienten mit Faktor-IX-Inhibitoren ein erhöhtes Anaphylaxie-Risiko bei fortgesetzter Faktor-IX-Behandlung aufweisen können.
Aufgrund des Risikos allergischer Reaktionen nach Gabe von Faktor-IX-Präparaten sollen die ersten Anwendungen von Faktor IX nach Ermessen des behandelnden Arztes unter ärztlicher Beobachtung durchgeführt werden, so dass gegebenenfalls geeignete Möglichkeiten zur Behandlung allergischer Reaktionen vorhanden sind.
Thromboembolie
Wegen des möglichen Risikos thromboembolischer Komplikationen sollte bei der Anwendung von Faktor-IX-Produkten bei Patienten mit Lebererkrankungen, postoperativen Patienten, Neugeborenen oder Patienten mit Risiko für thrombotische Ereignisse oder einer Verbrauchskoagulopathie (disseminierten intravaskulären Gerinnung, DIC) eine klinische Überwachung mit geeigneten biologischen Testverfahren erfolgen, um Frühzeichen einer thrombotischen Komplikation oder einer Verbrauchskoagulopathie festzustellen. In den genannten Fällen muss der Nutzen einer Behandlung mit ALPROLIX gegen die Risiken dieser Komplikationen abgewogen werden.
Kontinuierliche Infusion
Die Sicherheit und Wirksamkeit der Anwendung von ALPROLIX als kontinuierliche Infusion ist nicht belegt (siehe auch «Dosierung/Anwendung»).
Kardiovaskuläre Ereignisse
Bei Patienten mit bestehenden kardiovaskulären Risikofaktoren kann eine Substitutionstherapie mit Faktor IX Produkten das kardiovaskuläre Risiko erhöhen.
Katheter-assoziierte Komplikationen
Wenn ein zentraler Venenkatheter (ZVK) erforderlich ist, sollte das Risiko von ZVK-assoziierten Komplikationen einschliesslich lokaler Infektionen, Bakteriämie und Thrombosen an der Einstichstelle des Katheters berücksichtigt werden.
Kinder und Jugendliche
Die aufgeführten Warnhinweise und Vorsichtsmassnahmen gelten gleichermassen für Erwachsene und Kinder.
Hinweise zu Hilfsstoffen
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Durchstechflasche, d.h. es ist nahezu «natriumfrei». Esenthält 0.3 mmol (oder 6.4 mg) Natrium je Durchstechflasche. Dies ist bei Patienten zu berücksichtigen, die auf eine natriumarme Ernährung achten müssen.
Interaktionen
Es wurde über keine Interaktionen von ALPROLIX mit anderen Arzneimitteln berichtet. Es wurden keine Interaktionsstudien durchgeführt.
Schwangerschaft/Stillzeit
Mit ALPROLIX wurden keine tierexperimentellen Reproduktionsstudien durchgeführt. Eine Studie zum Plazentatransfer bei Mäusen wurde durchgeführt (siehe «Präklinische Daten»). Da Hämophilie B bei Frauen nur selten auftritt, liegen keine Erfahrungen mit der Anwendung von Faktor IX während der Schwangerschaft und Stillzeit vor. Daher sollte Faktor IX während der Schwangerschaft und Stillzeit nur angewendet werden, wenn dies eindeutig angezeigt ist.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
ALPROLIX hat keinen Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen.
Unerwünschte Wirkungen
Zusammenfassung des Sicherheitsprofils
Im Zusammenhang mit Faktor-Substitutionstherapien sind in seltenen Fällen Überempfindlichkeitsreaktionen oder allergische Reaktionen möglich, die Angioödem, brennendes und stechendes Gefühl an der Infusionsstelle, Schüttelfrost, Hitzegefühl, generalisierte Urtikaria, Nesselsucht, Hypotonie, Lethargie, Übelkeit, Unruhe, Tachykardie, Engegefühl in der Brust, Kribbeln, Erbrechen, Giemen einschliessen können. In einigen Fällen entwickelten sich diese Reaktionen zu einer schweren Anaphylaxie (einschliesslich Schock) und traten in engem zeitlichem Zusammenhang mit der Bildung von Faktor-IX-Inhibitoren auf (siehe auch Abschnitt „Warnhinweise und Vorsichtsmassnahmen”). Bei Patienten mit Hämophilie B können sich neutralisierende Antikörper (Inhibitoren) gegen Faktor IX bilden. Falls es zur Entwicklung solcher Inhibitoren kommt, äussert sich dies in einem unzureichenden klinischen Ansprechen. In solchen Fällen sollte ein spezialisiertes Hämophiliezentrum kontaktiert werden. Bei Versuchen die Immuntoleranz von Patienten zu induzieren, welche Hämophilie B mit Faktor-IX-Inhibitoren Bildung haben und allergische Reaktioenen zeigten, wurde von nephrotischem Syndrom berichtet.
Nach der Verabreichung von Faktor-IX-Präparaten besteht ein potenzielles Risiko thromboembolischer Episoden, wobei das Risiko bei Produkten von geringerem Reinheitsgrad höher ist. Bei Anwendung von hochreinem Faktor IX sind thromboembolische Komplikationen jedoch selten.
Tabellarische Auflistung der Nebenwirkungen
Vorbehandelte Patienten (PTPs): Die Sicherheit von ALPROLIX wurde von 153 Patienten mit schwerer Hämophilie B in zwei abgeschlossenen Phase-3-Studien sowie in einer Erweiterungsstudie beurteilt. Unerwünschte Wirkungen wurden über insgesamt 561 Patientenjahren überwacht. Die Gesamtanzahl an Expositionstagen betrug 26106 mit einer medianen Anzahl von 165 (Interval:1 bis 528) Expositionstagen pro Patient.
Zuvor unbehandelte Patienten (PUPs): Insgesamt 33 Patienten mit schwerer Hämophilie B wurden in einer klinischen Studie beobachtet. Unerwünschte Ereignisse wurden über insgesamt 57.51 Patientenjahre überwacht. Die Anzahl der Expositionstage betrug insgesamt 2233 mit einem Medianwert von 76 (Intervall 1 bis 137) Expositionstagen pro Patient.
Die unter ALPROLIX am häufigsten gemeldeten unerwünschten Wirkungen waren mit einer Inzidenz von 1,3% Kopfschmerzen, orale Parästhesie, obstruktive Uropathie (PTPs) und Erythem an der Injektionsstelle, sowie Faktor-IX-Inhibition und Überempfindlichkeit bei PUPs.
Die unerwünschten Wirkungen sind entsprechend der MedDRA-Systemorganklassifikation aufgeführt (SOC und bevorzugte Terms).
Die Häufigkeiten wurden gemäss folgender Konvention beurteilt: sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1000, <1/100); selten (≥1/10'000, <1/1000); sehr selten (<1/10'000), /Einzelfälle (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). In der Tabelle sind Nebenwirkungen aufgeführt, die in klinischen Studien gemeldet bzw. im Rahmen der Anwendungsbeobachtung (nach Markteinführung) festgestellt wurden.
Tabelle 2: Nebenwirkungen von ALPROLIX
Systemorganklasse gemäss MedDRA | Nebenwirkungen | Häufigkeitskategorie |
Erkrankungen des Blutes und des Lymphsystems | Faktor-IX-Inhibition | Häufig1 |
Erkrankungen des Immunsystems | Überempfindlichkeit Anaphylaktische Reaktion | Häufig1 Einzelfälle |
Stoffwechsel- und Ernährungsstörungen | Verminderter Appetit | Gelegentlich |
Erkrankungen des Nervensystems | Kopfschmerzen Schwindelgefühl Geschmacksstörung | Häufig Gelegentlich Gelegentlich |
Herzerkrankungen | Palpitationen | Gelegentlich |
Gefäßerkrankungen | Hypotonie | Gelegentlich |
Erkrankungen des Gastrointestinaltrakts | orale Parästhesie Mundgeruch | Häufig Gelegentlich |
Erkrankungen der Nieren und Harnwege | obstruktive Uropathie Hämaturie Nierenkolik Nephrotisches Syndrom (nach versuchter Immuntoleranzinduktion) | Häufig Gelegentlich Gelegentlich Einzelfälle |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | Erythem an der Injektionsstelle Ermüdung Schmerzen an der Infusionsstelle | Häufig Gelegentlich Gelegentlich |
1 Die Häufigkeit bezieht sich auf das Auftreten in der Studie mit PUPs. Die beiden Ereignisse Faktor-IX-Inhibition und Überempfindlichkeit traten in Studie IV bei demselben PUP auf. Siehe Beschreibung ausgewählter Nebenwirkungen.
Beschreibung ausgewählter Nebenwirkungen
Im gesamten klinischen Studienprogramm trat bei einem (zuvor unbehandelten) Patienten in Studie IV ein niedriger Titer von Faktor-IX-Hemmkörpern in Verbindung mit Überempfindlichkeit auf (siehe «Klinische Wirksamkeit»). Nach der Markteinführung wurden die Entwicklung von Faktor-IX-Inhibitoren und Überempfindlichkeit (einschließlich Anaphylaxie) beobachtet.
Kinder und Jugendliche
Bei Kindern sind unerwünschte Wirkungen derselben Häufigkeit, Art und Schwere zu erwarten wie bei Erwachsenen. Informationen zum Umfang und der Altersstruktur der Sicherheitsdatenbank bei Kindern siehe «Pharmakokinetik»).
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Es wurden keine Fälle von Überdosierung berichtet.
Eigenschaften/Wirkungen
ATC-Code
B02BD04
Wirkungsmechanismus
Eftrenonacog alfa (rekombinanter humaner Gerinnungsfaktor IX, Fc-Fusionsprotein (rFIXFc)) besitzt 867 Aminosäuren. Es wird mittels rekombinanter DNA-Technologie in einer humanen embryonalen Nieren-Zelllinie (HEK) und ohne Zugabe von exogenen menschlichen oder tierischen Proteinen im Zellkulturprozess, in der Aufreinigung oder in der Endformulierung hergestellt.
Faktor IX ist ein einkettiges Glykoprotein mit einer Molekülmasse von etwa 55000 Dalton. Der Gerinnungsfaktor ist Vitamin-K-abhängig. Faktor IX wird durch Faktor XIa im endogenen Gerinnungssystem und durch Faktor VII/Gewebefaktorkomplex im exogenen Gerinnungssystem aktiviert. In Verbindung mit aktiviertem Faktor VIII aktiviert der aktivierte Faktor IX den Faktor X. Der aktivierte Faktor X wandelt Prothrombin in Thrombin um. Thrombin wandelt dann Fibrinogen in Fibrin um und es bildet sich ein Blutgerinnsel.
Hämophilie B ist eine geschlechtsgebundene erbliche Störung der Blutgerinnung, bedingt durch niedrige Spiegel des funktionellen Faktors IX. Sie führt zu Blutungen in Gelenken, Muskeln oder inneren Organen, entweder spontan oder infolge eines unfallbedingten oder chirurgischen Traumas. Durch Substitutionstherapie wird der Faktor-IX-Spiegel im Plasma angehoben, wodurch der Faktor-IX-Mangel und die Blutungstendenz vorübergehend korrigiert werden können.
Pharmakodynamik
ALPROLIX (Eftrenonacog alfa) ist ein langwirkendes, vollständig rekombinantes Fusionsprotein, bestehend aus humanem Gerinnungsfaktor IX, der kovalent an die Fc-Domäne des humanen Immunglobulins G1 gebunden ist, und wird mittels rekombinanter DNA-Technologie hergestellt.
Die Fc-Region des humanen Immunglobulins G1 bindet an den neonatalen Fc-Rezeptor. Dieser Rezeptor wird während des gesamten Lebens exprimiert und bildet Teil eines natürlichen Stoffwechselwegs, der Immunglobuline vor dem lysosomalen Abbau schützt, indem er diese Proteine in den Kreislauf zurückführt, was zu einer langen Halbwertszeit im Plasma führt.
Klinische Wirksamkeit
Die Sicherheit, Wirksamkeit und Pharmakokinetik von ALPROLIX wurden in zwei multinationalen, offenen Zulassungsstudien bei vorbehandelten Patienten (PTPs) - einer Phase-3-Studie (Studie I mit Erwachsenen und Jugendlichen) und einer pädiatrischen Phase-3-Studie (Studie II) - beurteilt (siehe «Kinder und Jugendliche»). Die Sicherheit und Wirksamkeit von ALPROLIX wurden ausserdem bei zuvor unbehandelten Patienten (PUPs) mit schwerer Hämophilie B untersucht (Studie IV); siehe «Kinder und Jugendliche».
In Studie I erfolgte ein Vergleich der Wirksamkeit zweier prophylaktischer Behandlungsregime (fixes wöchentliches Intervall mit einer Dosis von 50 I.E./kg und individualisiertes Dosierungsintervall mit 100 I.E./kg beginnend alle 10 Tage) mit der einer Bedarfsbehandlung. In die Studie wurden insgesamt 123 vorbehandelte männliche Patienten (12 bis 71 Jahre) mit schwerer Hämophilie B (endogene FIX-Aktivität ≤2%) eingeschlossen. Alle Patienten erhielten ALPROLIX und wurden bis zu 77 Wochen nachbeobachtet.
Von den 123 Patienten, die Studie I abschlossen, wurden 93 in die Erweiterungsstudie (Studie III) mit einem Nachbeobachtungszeitraum vom 6,5 Jahren aufgenommen.
Es sollte beachtet werden, dass die annualisierten Blutungsraten (Annualised Bleeding Rates, ABR) nicht vergleichbar sind zwischen verschiedenen Faktor-Konzentraten und zwischen verschiedenen klinischen Studien.
Prophylaxe mit festem wöchentlichem und individualisiertem Dosierungsintervall:
Die mediane Wochendosis betrug bei Patienten im Arm mit festem wöchentlichen Dosierungsintervall der Studie I 45,17 I.E./kg (Interquartilsabstand [Interquartile Range, IQR] 38,1-53,7). Die entsprechenden medianen ABR betrugen bei den hinsichtlich der Wirksamkeit auswertbaren Patienten 2,95 (IQR: 1,01-4,35) und blieben im Verlauf der Studie III ähnlich (1,85 [IQR: 0,76-4,0]). Die Patienten in Studie III wiesen eine mediane Anzahl von 0,38 (IQR: 0,00-1,43) spontanen Gelenkblutungen auf.
Bei den Patienten im Arm mit individualisiertem Dosierungsintervall der Studie I betrug das mediane Dosierungsintervall 12,53 Tage (IQR: 10,4-13,4). Die entsprechende mediane ABR betrug 1,38 (IQR: 0,00-3,43) und blieb im Verlauf der Studie III ähnlich (1,85 [IQR: 0,76-4,0]).
Bei beiden prophylaktischen Behandlungsregimes waren Dosierungsintervalle und Faktorverbrauch in Studie III (Erweiterungsstudie) ähnlich wie in Studie I.
Bei 42 % der Patienten unter individualisierter Prophylaxe und 23,0 % der Patienten unter wöchentlicher Prophylaxe traten keine Blutungsepisoden auf. Der Anteil an Patienten mit ≥1 Zielgelenk zu Studienbeginn war bei der Prophylaxe mit individualisiertem Dosierungsintervall geringer als bei der wöchentlichen Prophylaxe (27,6 % bzw. 57,1 %).
Behandlung von Blutungen:
Von den 636 Blutungsereignissen, die in Studie I beobachtet wurden, konnten 90.4% mit 1 Injektion und insgesamt 97,3% mit 2 oder weniger Injektionen unter Kontrolle gebracht werden. Die mediane Dosis je Injektion zur Behandlung einer Blutungsepisode betrug 46,07 I.E./kg (IQR 32,86-57,03). Die mediane Gesamtdosis zur Behandlung einer Blutungsepisode betrug 51,47 I.E./kg (IQR 35,21- 61,73). im Behandlungsarm mit wöchentlicher Prophylaxe, 49,62 I.E./kg (35,71-94,82) im Behandlungsarm mit individualisierter Prophylaxe und 46,58 I.E./kg (33,33 -59,41) im Behandlungsarm unter Bedarfsbehandlung.
Perioperatives Management (chirurgische Prophylaxe):
Insgesamt wurden in Studie I und Studie III 35 grössere chirurgische Eingriffe vorgenommen und bei 22 Patienten (21 Erwachsene und Jugendliche und 1 Kind <12 Jahre) beurteilt. Bei 28 (80,0%) der 35 grösseren chirurgischen Eingriffe war vor der Operation die Gabe einer Einzeldosis zur Aufrechterhaltung der Hämostase während der Operation erforderlich. Die mediane Durchschnittsdosis pro Injektion zur Aufrechterhaltung der Hämostase während der Operation betrug 94,7 I.E./kg (Bereich: 49 bis 152 I.E./kg). Die Gesamtdosis am Operationstag reichte von 49 bis 341 I.E./kg und die Gesamtdosis im 14-tägigen perioperativen Zeitraum reichte von 60 bis 1947 I.E./kg.
Das hämostatische Ansprechen wurde bei 100% der grösseren chirurgischen Eingriffe als hervorragend oder gut beurteilt.
Kinder und Jugendliche
In Studie II wurden insgesamt 30 vorbehandelte männliche pädiatrische Patienten mit schwerer Hämophilie B (endogene FIX-Aktivität ≤2%) aufgenommen. Die Patienten waren unter 12 Jahre alt (15 waren <6 Jahre alt und 15 waren 6 bis <12 Jahre alt). Alle Patienten erhielten ALPROLIX und wurden bis zu 52 Wochen nachbeobachtet.
Alle 30 Patienten erhielten ALPROLIX im Rahmen eines prophylaktischen Behandlungsregimes, beginnend mit 50-60 I.E./kg alle 7 Tage, mit Dosisanpassungen bis maximal 100 I.E./kg und einem Dosierungsintervall von mindestens einmal und maximal zweimal wöchentlich.
Von den 30 Patienten, die Studie II abschlossen, wurden 27 in Studie III (Erweiterungsstudie) aufgenommen. Die mediane Verweildauer in Studie II+III betrug 2,88 Jahre mit einer medianen Anzahl von 166 Expositionstagen.
In Studie IV wurden 33 zuvor unbehandelte Patienten (PUPs) im Kindes- und Jugendalter mit schwerer Hämophilie B (≤2 % endogene FIX-Aktivität) eingeschlossen. Das mediane Alter bei Einschluss in die Studie war 0,6 Jahre (Bereich: 0,08 bis 2 Jahre); 78,8 % der Teilnehmer waren unter 1 Jahr alt. Die mediane Dauer der Behandlung mit ALPROLIX betrug insgesamt 83,01 Wochen (Bereich: 6,7 bis 226,7 Wochen), mit einer medianen Anzahl von insgesamt 76 Expositionstagen (Bereich: 1 bis 137 Tage).
Prophylaxe mit individualisiertem Behandlungsregime:
In Studie II betrug die mediane durchschnittliche Wochendosis von ALPROLIX 59,40 I.E./kg (Interquartilsabstand 52,95 bis 64,78 I.E./kg) für Patienten < 6 Jahre und 57,78 I.E./kg (Interquartilsabstand 51,67 bis 65,01 I.E./kg) für Patienten im Alter von 6 bis < 12 Jahren. Das mediane Dosierungsintervall betrug insgesamt 6,99 Tage (Interquartilsabstand 6,94 bis 7,03). Es gab keine Unterschiede zwischen den medianen Dosierungsintervallen der beiden Altersgruppen. Mit Ausnahme eines Patienten, dessen letzte verschriebene Dosis 100 I.E./kg alle 5 Tage betrug, lagen die letzten verschriebenen Dosen bei den anderen 29 Patienten bei bis zu 70 I.E./kg alle 7 Tage. Bei 33 % der pädiatrischen Patienten traten keine Blutungsepisoden auf. Dosierungsintervalle und Faktorverbrauch waren in Studie III ähnlich wie in Studie II.
Die medianen annualisierten Blutungsraten bei hinsichtlich der Wirksamkeit auswertbaren Patienten unter 12 Jahren der Studie II betrugen 1,97 (Interquartilsabstand 0,00 bis 3,13) und blieben im Verlauf der Studie III (Erweiterungsstudie) ähnlich.
Bei den PUPs (Studie IV) betrug die mediane durchschnittliche Wochendosis ALPROLIX 57,96 I.E./kg (Interquartilsabstand 52,45 bis 65,06 I.E./kg), und das mediane Dosierungsintervall betrug 7 Tage (Interquartilsabstand 6,95 bis 7,12 Tage). Dosierungsintervalle und Faktorverbrauch waren in Studie IV ähnlich wie in Studie II und III. Von den PUPs, die eine Prophylaxe erhielten, hatten 8 (28,6 %) keine Blutungsepisoden. Die mediane ABR von allen Teilnehmern unter Prophylaxe betrug 1,24 (Interquartilsabstand 0,0 bis 2,49).
Behandlung von Blutungsepisoden:
Von den 60 Blutungsereignissen, die in Studie II beobachtet wurden, konnten 75% mit 1 Injektion und insgesamt 91,7% mit 2 oder weniger Injektionen unter Kontrolle gebracht werden. Die mediane Dosis je Injektion zur Behandlung einer Blutungsepisode betrug 63,51 I.E./kg (Interquartilbereich 48,92 bis 99,44). Die gesamte mediane Dosis zur Behandlung einer Blutungsepisode betrug 68,22 I.E./kg (Interquartilbereich 50,89 bis 126,19).
Von den 58 Blutungsereignissen bei PUPs unter Prophylaxe in Studie IV konnten 87,9 % mit 1 Injektion unter Kontrolle gebracht werden und insgesamt 96,6 % mit 2 Injektionen oder weniger. Die mediane durchschnittliche Dosis pro Injektion zur Behandlung einer Blutungsepisode betrug 71,92 I.E./kg (Interquartilsabstand 52,45 bis 100,81 I.E./kg). Die mediane Gesamtdosis zur Behandlung einer Blutungsepisode betrug 78,74 I.E./kg (Interquartilsabstand 53,57 bis 104,90 I.E./kg).
Pharmakokinetik
Absorption
Alle Studien zur Pharmakokinetik von ALPROLIX wurden an vorbehandelten Patienten mit schwerer Hämophilie B durchgeführt. Die in diesem Abschnitt vorgestellten Daten wurden mittels Einstufen-Gerinnungstest mit einem gegen einen Faktor-IX-Plasmastandard kalibrierten siliciumbasierten aPTT-Reagenz erhoben. Die pharmakokinetischen Eigenschaften wurden bei 22 Patienten (≥19 Jahre) unter Behandlung mit ALPROLIX (rFIXFc) beurteilt. Die Entnahme der Pharmakokinetik-Proben erfolgte vor der Verabreichung von 50 I.E./kg rFIX (Benefix) sowie zu 8 Zeitpunkten über einen Zeitraum von bis zu 96 Stunden (4 Tagen) nach der Verabreichung. Nach einer Auswaschphase von mindestens 120 Stunden (5 Tagen) erhielten die Patienten dann eine Einzeldosis von 50 I.E./kg ALPROLIX. Die Entnahme der Pharmakokinetik-Proben erfolgte vor der Verabreichung sowie anschliessend zu 11 Zeitpunkten über einen Zeitraum von bis zu 240 Stunden (10 Tagen) nach der Verabreichung. Die pharmakokinetischen Parameter nach Gabe einer Dosis von 50 I.E./kg ALPROLIX sind in Tabelle 3 dargestellt.
Tabelle 3: Pharmakokinetische Parameter von ALPROLIX (50 I.E./kg)
Pharmakokinetische Parameter1 | ALPROLIX |
N=22 | |
Inkrementelle Recovery (I.E./dl je I.E./kg) | 0,92 |
AUC/Dosis (I.E.*h/dl je I.E./kg) | 31.32 |
Cmax (I.E./dl) | 40,81 |
CL (ml/h/kg) | 3,19 |
t1/2α (h) | 5,03 |
t1/2β (h) | 82,12 |
MRT (h) | 98,60 |
Vss (ml/kg) | 314,80 |
Zeit bis 1% (Tage) | 11,22 (10,20-12,35) |
1 Die pharmakokinetischen Parameter sind als geometrische Mittelwerte (95%-KI) dargestellt
Abkürzungen: KI = Konfidenzintervall; Cmax = maximale Aktivität; AUC = Fläche unter der
FIX-Aktivitäts-/Zeit-Kurve; t1/2α = Verteilungshalbwertszeit;
t1/2β = Eliminationshalbwertszeit; CL = Clearance; Vss = Verteilungsvolumen im Steady State;
MRT = mittlere Verweildauer.
Die Eliminationshalbwertszeit von ALPROLIX (82 Stunden) wird von der Fc-Region beeinflusst, die in Tiermodellen erwiesenermassen durch den neonatalen Fc-Rezeptor-Stoffwechselweg vermittelt wird.
Es wurde ein Populationspharmakokinetik-Modell entwickelt, das auf den Daten zur FIX-Aktivität von 161 Patienten jeglichen Alters (2-76 Jahre) mit einem Gewicht zwischen 12.5 und 186.7 kg beruhte, die an drei klinischen Studien teilgenommen hatten (12 Patienten aus einer Phase-1/2a-Studie sowie 123 Patienten aus Studie I und 26 Patienten aus Studie II). Die Erwartung der Clearance von ALPROLIX für einen typischen 70 kg schweren Erwachsenen ist 2.30 dl/h und das Verteilungsvolumen im Steady State von ALPROLIX ist 194.8 dl. Das Modell wurde für die Vorhersage des Aktivitäts-/Zeit-Profils nach einer Einzeldosis ALPROLIX bei Patienten mit schwerer Hämophilie B verwendet (siehe Tabelle 4).
Tabelle 4: Vorhergesagte FIX-Aktivität [I.E./dl] nach einer Einzeldosis ALPROLIX1 für Patienten im Alter von ≥12 Jahren
Dosis (I.E./kg) | Ende der Injektion | 12 Stunden | 24 Stunden (Tag 1) | 36 Stunden | 48 Stunden (Tag 2) | 72 Stunden (Tag 3) | Tag 5 | Tag 7 | Tag 10 | Tag 14 |
Median (5., 95.) | ||||||||||
50 | 52,0 | 21,7 | 15,2 | 11,3 | 8,4 | 5,5 | 3,02 | 1,93 | 1,07 | 0,495 |
[31,8; 82] | [15,0; 31,0] | [10,2; 21,5] | [7,50; 16,5] | [5,5; 12,5] | [3,59; 8,25] | [1,88; 4,65] | [0.99; 3.12] | [0,345; 1,95] | [0,0829; 1,17] | |
100 | 104,0 | 43,4 | 30,4 | 22,6 | 16,8 | 11 | 6,03 | 3,85 | 2,13 | 0,991 |
[63,6; 164] | [30,0; 62,0] | [20,4; 42,9] | [15,0; 32,9] | [11; 24,9] | [7,18; 16,5] | [3,77; 9,29 | [1,98; 6,24] | [0,691; 3,89] | [0,166; 2,34] | |
1 Siehe „Dosierung/Anwendung“
Distribution
Siehe unter „Absorption”.
Metabolismus
Siehe unter „Absorption”.
Elimination
Siehe unter „Absorption”.
Kinetik spezieller Patientengruppen
Kinder und Jugendliche
Die pharmakokinetischen Parameter von ALPROLIX wurden bei Jugendlichen in Studie I und bei Kindern in Studie II ermittelt (die Entnahme der Pharmakokinetik-Proben erfolgte vor der Verabreichung sowie zu mehreren Zeitpunkten über einen Zeitraum von bis zu 336 Stunden (14 Tagen) nach der Verabreichung in Studie I bzw. vor der Verabreichung sowie zu 7 Zeitpunkten über einen Zeitraum von bis zu 168 Stunden (7 Tagen) nach der Verabreichung in Studie II).
Tabelle 5 zeigt die pharmakokinetischen Parameter, die anhand der pädiatrischen Daten von 35 Patienten unter 18 Jahren berechnet wurden.
Tabelle 5: Vergleich der pharmakokinetischen Parameter von ALPROLIX (rFIXFc) nach Alterskategorie
Pharmakokinetische Parameter1 | Studie II | Studie I | |
<6 Jahre (Bereich 2-4) | 6 bis <12 Jahre (Bereich 6-10) | 12 bis <18 Jahre (Bereich 12-17) | |
N = 11 | N = 13 | N = 11 | |
IR (I.E./dl je I.E./kg) | 0,5898 | 0,7170 | 0,8470 |
AUC/Dosis (I.E.*h/dl je I.E./kg) | 22,71 | 28,53 | 29,50 |
t½ (h) | 66,49 | 70,34 | 82,22 |
MRT (h) | 83,65 | 82,46 | 93,46 |
CL (ml/h/kg) | 4,365 | 3,505 | 3,390 |
Vss (ml/kg) | 365,1 | 289,0 | 316,8 |
1 Die pharmakokinetischen Parameter aus der nicht-kompartimentellen Analyse sind als geometrische Mittelwerte (95%-KI) dargestellt Abkürzungen: KI = Konfidenzintervall; IR = Inkrementelle Recovery; AUC = Fläche unter der FIX-Aktivitäts-/Zeit-Kurve; t1/2 = terminale Halbwertszeit; MRT = mittlere Verweildauer; CL = Clearance; Vss = Verteilungsvolumen im Steady State | |||
Präklinische Daten
Basierend auf den Studien zur akuten Toxizität und Toxizität bei wiederholter Gabe, die die Beurteilungen der lokalen Toxizität und der Sicherheitspharmakologie beinhalteten, lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen. Studien zur Untersuchung der Genotoxizität, Kanzerogenität, Reproduktionstoxizität oder embryonalen/fetalen Entwicklung wurden nicht durchgeführt. In einer Studie zum Plazentatransfer wurde gezeigt, dass Eftrenonacog alfa (rFIXFc) bei Mäusen in geringen Konzentrationen die Plazentaschranke passiert.
Fertilität
Mit ALPROLIX wurden keine tierexperimentellen Fertilitätsstudien durchgeführt.
Sonstige Hinweise
Inkompatibilitäten
Da keine Verträglichkeitsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Es sollte ausschliesslich das mitgelieferte Infusionsset verwendet werden, da es bei manchen Injektionsausrüstungen infolge einer Adsorption von Gerinnungsfaktor IX an den inneren Oberflächen zu einem Behandlungsversagen kommen kann.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden. Während der Dauer der Haltbarkeit kann das Arzneimittel für einen einmaligen Zeitraum von höchstens 6 Monaten bei Raumtemperatur (bis zu 30°C) gelagert werden. Der Tag, an dem das Arzneimittel aus dem Kühlschrank genommen wird, soll auf dem Karton vermerkt werden. Nachdem das Arzneimittel bei Raumtemperatur aufbewahrt wurde, darf es nicht wieder in den Kühlschrank gestellt werden.
Nach der Rekonstitution
Die chemische und physikalische Stabilität wurde für 6 Stunden bei Raumtemperatur (bis zu 30°C) gezeigt. Wenn das Arzneimittel nicht innerhalb von 6 Stunden verwendet wird, muss es verworfen werden. Aus mikrobiologischer Sicht sollte das Produkt unmittelbar nach Rekonstitution verwendet werden. Falls dies nicht möglich ist, liegen Aufbrauchfristen und Lagerbedingungen in der Verantwortung des Anwenders. Das Arzneimittel vor direktem Sonnenlicht schützen.
Besondere Lagerungshinweise
Arzneimittel für Kinder unzugänglich aufbewahren.
Die Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen. Im Kühlschrank (2-8°C) lagern. Nicht einfrieren. Dieses Arzneimittel ist für den Einmalgebrauch bestimmt. Die Restlösung ist sachgerecht zu entsorgen.
Aufbewahrungsbedingungen nach Rekonstitution des Arzneimittels siehe «Haltbarkeit».
Hinweise für die Handhabung
Der Inhalt der Durchstechflasche mit dem lyophilisierten Pulver zur Herstellung einer Injektionslösung muss mit dem mitgelieferten Lösungsmittel (Natriumchloridlösung) in der Fertigspritze unter Verwendung des sterilen Durchstechflaschenadapters rekonstituiert werden. Die Durchstechflasche solltevorsichtig geschwenkt werden, bis das Pulver vollständig gelöst ist.
Die rekonstituierte Lösung sollte klar bis schwach schillernd und farblos sein. Das rekonstituierte Arzneimittel sollte vor der Verabreichung visuell auf Partikel und Verfärbungen untersucht werden. Die Lösung darf nicht verwendet werden, wenn sie trübe ist oder Ablagerungen aufweist.
Dieses Produkt ist nur zum einmaligen Gebrauch. ALPROLIX sollte nicht mit anderen Injektions- oder Infusionslösungen gemischt werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial sind sachgemäss zu entsorgen.
Eine ALPROLIX-Packung enthält:
|
|

Vorbereitung:
Vor dem Öffnen der Packung Hände waschen.
1. Kontrollieren Sie die auf der Packung angegebene Bezeichnung und Dosierungsstärke, um sicherzustellen, dass sie das richtige Produkt enthält. Überprüfen Sie das Verfalldatum auf dem Umkarton von ALPROLIX. Nicht verwenden, wenn das Verfalldatum des Arzneimittels überschritten ist. | |
2. Wenn ALPROLIX in einem Kühlschrank aufbewahrt wurde, warten Sie vor der Anwendung, bis die Durchstechflasche mit ALPROLIX (A) und die Spritze mit dem Lösungsmittel (B) Raumtemperatur angenommen haben. Verwenden Sie keine externe Wärmequelle. | |
3. Stellen Sie die Durchstechflasche auf eine saubere, ebene Oberfläche. Entfernen Sie die Flip-Top-Kappe aus Kunststoff von der Durchstechflasche. |
|
4. Reinigen Sie die Oberseite der Durchstechflasche mit einem der in der Packung enthaltenen Alkoholtupfer (F) und lassen Sie sie an der Luft trocknen. Vermeiden Sie nach dem Abwischen jede Berührung der Oberseite der Durchstechflasche bzw. einen Kontakt der Oberseite mit anderen Gegenständen. |
|
5. Ziehen Sie das Schutzpapier vom durchsichtigen Kunststoffadapter (D) ab. Nehmen Sie den Adapter nicht aus der Schutzkappe heraus. Vermeiden Sie es, die Innenseite der Packung des Durchstechflaschen-Adapters zu berühren. | |
6.Stellen Sie die Durchstechflasche auf eine ebene Oberfläche. Halten Sie den Durchstechflaschenadapter an seiner Schutzkappe fest und setzen Sie ihn gerade auf die Oberseite der Durchstechflasche. Drücken Sie den Adapter fest nach unten, bis er oben auf der Durchstechflasche einrastet und der Adapterdorn durch den Stopfen der Durchstechflasche dringt. |
|
7. Verbinden Sie die Kolbenstange (C) mit der Lösungsmittelspritze, indem Sie die Spitze der Kolbenstange in die Öffnung des Spritzenkolbens einführen. Drehen Sie die Kolbenstange kräftig im Uhrzeigersinn, bis sie fest und sicher im Spritzenkolben sitzt. |
|
8. Brechen Sie die weisse, manipulationssichere Kunststoffkappe von der Lösungsmittel-Spritze ab, indem Sie sie an der Perforation nach unten biegen, bis sie bricht. Legen Sie die Kappe mit der Oberseite nach unten auf eine ebene Oberfläche. Berühren Sie nicht die Innenseite der Kappe oder die Spritzenspitze. |
|
9. Entfernen Sie die Schutzkappe vom Adapter und entsorgen Sie sie. |
|
10. Verbinden Sie die Lösungsmittelspritze mit dem Durchstechflaschenadapter, indem Sie die Spitze der Spritze in die Adapteröffnung einführen. Drücken und drehen Sie die Spritze kräftig im Uhrzeigersinn, bis sie fest und sicher sitzt. |
|
11. Drücken Sie die Kolbenstange langsam nach unten, um das gesamte Lösungsmittel in die Durchstechflasche mit ALPROLIX zu injizieren. |
|
12. Lassen Sie die Spritze am Adapter und die Kolbenstange heruntergedrückt und schwenken Sie die Durchstechflasche vorsichtig, bis sich das Pulver gelöst hat. Nicht schütteln. |
|
13. Die fertige Lösung muss vor der Verabreichung visuell überprüft werden. Die Lösung soll klar bis schwach schillernd und farblos sein. Sie darf nicht verwendet werden, wenn sie trübe ist oder sichtbare Partikel enthält. | |
14. Achten Sie darauf, dass die Kolbenstange in der Spritze weiterhin vollständig heruntergedrückt ist, und drehen Sie dann die Durchstechflasche auf den Kopf. Ziehen Sie die Kolbenstange langsam zurück, um die gesamte Lösung durch den Durchstechflaschenadapter in die Spritze aufzuziehen. Hinweis: Wenn Sie mehr als eine Durchstechflasche von ALPROLIX pro Injektion verwenden, bereiten Sie jede Durchstechflasche gemäss den obigen Anweisungen (Schritte 1 bis 13) einzeln zu. Entfernen Sie die Lösungsmittel-Spritze und lassen Sie den Durchstechflaschenadapter angeschlossen. Zum Aufziehen der zubereiteten Lösungen aus jeder der einzelnen Durchstechflaschen kann eine einzelne grosse Luer-Lock-Spritze verwendet werden. |
|
15. Entfernen Sie die Spritze vom Durchstechflaschenadapter, indem Sie die Durchstechflasche vorsichtig ziehen und gegen den Uhrzeigersinn drehen. |
|
16. Entsorgen Sie die Durchstechflasche und den Adapter. Hinweis: Wenn Sie die Lösung nicht sofort anwenden, sollte die Spritzenkappe vorsichtig wieder auf die Spritzenspitze aufgesetzt werden. Berühren Sie nicht die Spritzenspitze oder die Innenseite der Kappe. Nach der Zubereitung kann ALPROLIX vor der Verabreichung bis zu 6 Stunden bei Raumtemperatur aufbewahrt werden. Nach dieser Zeit muss das zubereitete ALPROLIX entsorgt werden. Vor direkter Sonneneinstrahlung schützen. | |











Verabreichung:
1.Öffnen Sie die Packung mit dem Infusionsset und entfernen Sie die Kappe am Ende des Schlauchs. Schließen Sie die Spritze mit der zubereiteten ALPROLIX-Lösung durch Drehen im Uhrzeigersinn an das Ende des Infusionsschlauchs an. |
|
2.Verwenden Sie bei Bedarf einen Stauschlauch (Tourniquet) und bereiten Sie die Injektionsstelle vor, indem Sie die Haut gründlich mit dem zweiten Alkoholtupfer aus der Packung abwischen.
| |
3. Entfernen Sie die gesamte Luft aus dem Infusionsschlauch, indem Sie die Kolbenstange langsam herunterdrücken, bis Flüssigkeit die Nadel des Infusionsbestecks erreicht hat. Drücken Sie die Lösung nicht durch die Nadel. Nehmen Sie die durchsichtige Kunststoffschutzhülle von der Nadel ab. | |
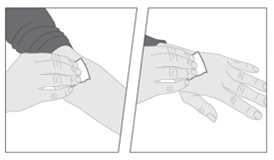
4. Führen Sie die Nadel des Infusionsbestecks, wie von ihrem Arzt oder dem medizinischen Fachpersonal gezeigt, in eine Vene ein und entfernen Sie das Tourniquet. Wenn Sie möchten, können Sie eines der Pflaster (G) aus der Packung verwenden, um die Kunststoffflügel der Nadel an der Injektionsstelle zu fixieren. Das zubereitete Arzneimittel soll über mehrere Minuten intravenös injiziert werden. Möglicherweise wird Ihr Arzt die für Sie empfohlene Injektionsgeschwindigkeit ändern, damit es für Sie angenehmer ist. | |
5. Nachdem die Injektion beendet und die Nadel entfernt wurde, klappen Sie den Nadelschutz über die Nadel und lassen ihn einrasten. |
|
6.Entsorgen Sie die gebrauchte Nadel, nicht verwendete Lösung, die Spritze und die leere Durchstechflasche auf sichere Weise in einem geeigneten Behälter für medizinische Abfälle, da diese Materialien andere Menschen verletzen können, wenn sie nicht ordnungsgemäss beseitigt werden. Die Ausrüstung darf nicht wiederverwendet werden. | |


Zulassungsnummer
66039 (Swissmedic)
Zulassungsinhaberin
Swedish Orphan Biovitrum AG, Basel
Stand der Information
Dezember 2021
Composizione
Principi attivi
Eftrenonacog alfa (fattore IX della coagulazione umano (rDNA))
Sostanze ausiliarie
Polvere: saccarosio, istidina, mannitolo, polisorbato 20, sodio idrossido (per la regolazione del pH), acido cloridrico (per la regolazione del pH)
Solvente: soluzione di sodio cloruro, acqua per preparazioni iniettabili
Sodio per flaconcino: 0,3 mmol (6,4 mg)
Forma farmaceutica e quantità di principio attivo per unità
Polvere e solvente per soluzione iniettabile (e.v.).
1 flaconcino di polvere ALPROLIX contiene eftrenonacog alfa nelle seguenti concentrazioni:
250 UI di eftrenonacog alfa (50 UI/ml dopo ricostituzione con 5 ml di solvente (soluzione di sodio cloruro))
500 UI di eftrenonacog alfa (100 UI/ml dopo ricostituzione con 5 ml di solvente (soluzione di sodio cloruro))
1000 UI di eftrenonacog alfa (200 UI/ml dopo ricostituzione con 5 ml di solvente (soluzione di sodio cloruro))
2000 UI di eftrenonacog alfa (400 UI/ml dopo ricostituzione con 5 ml di solvente (soluzione di sodio cloruro))
3000 UI di eftrenonacog alfa (600 UI/ml dopo ricostituzione con 5 ml di solvente (soluzione di sodio cloruro))
Indicazioni/Possibilità d'impiego
ALPROLIX è indicato per il trattamento e la profilassi delle emorragie in pazienti con emofilia B (deficit congenito di fattore IX).
Posologia/Impiego
Il trattamento deve essere iniziato sotto la supervisione di un medico esperto nel trattamento dell'emofilia.
Monitoraggio della terapia
Durante la terapia, si raccomanda un'adeguata determinazione dei livelli di fattore IX per stabilire il dosaggio da somministrare e la frequenza con cui ripetere le iniezioni. La risposta dei singoli pazienti al fattore IX può variare, manifestandosi con differenti emivite e recuperi diversi. Il dosaggio basato sul peso corporeo può richiedere un adeguamento per i pazienti sottopeso o sovrappeso. È indispensabile uno stretto monitoraggio della terapia sostitutiva attraverso l'analisi della coagulazione (attività del fattore IX nel plasma), soprattutto durante gli interventi chirurgici maggiori.
Quando si utilizza un test di coagulazione one-stage in vitro basato sul tempo di tromboplastina (aPTT) per determinare l'attività del fattore IX nei campioni di sangue del paziente, i risultati dell'attività del fattore IX possono essere significativamente influenzati sia dal tipo di reagente aPTT sia dallo standard di riferimento utilizzati. Questo è particolarmente importante se si cambia il laboratorio e/o i reagenti usati nel test.
È probabile che le misurazioni effettuate con un test di coagulazione one-stage che utilizza un reagente aPTT a base di caolino portino a una sottostima del livello di attività.
Posologia
Il dosaggio e la durata della terapia sostitutiva dipendono dalla gravità del deficit di fattore IX, dalla sede e dall'entità dell'emorragia e dalle condizioni cliniche del paziente.
Il numero di unità di fattore IX da somministrare è espresso in unità internazionali (UI) sulla base dell'attuale standard dell'OMS per i preparati di fattore IX. L'attività del fattore IX nel plasma è espressa in percentuale (riferita al plasma umano normale) o in unità internazionali (riferite allo standard internazionale per il fattore IX plasmatico).
Un'unità internazionale (UI) di fattore IX Fc ricombinante equivale alla quantità di fattore IX in un ml di plasma umano normale.
Trattamento al bisogno
Il calcolo della dose necessaria di fattore IX Fc ricombinante si basa sul dato empirico per cui 1 unità internazionale (UI) di fattore IX per kg di peso corporeo aumenta l'attività del fattore IX nel plasma dell'1% dell'attività normale (UI/dl). La dose necessaria è calcolata con la seguente formula:
Unità richieste = peso corporeo (kg) x aumento desiderato del fattore IX (%) (UI/dl) x reciproco del recupero determinato (UI/kg per UI/dl).
La dose necessaria e la frequenza di somministrazione devono sempre essere orientate all'efficacia clinica nel singolo caso. Se è necessario ripetere la somministrazione per controllare l'emorragia, si deve tener conto dell'emivita prolungata di ALPROLIX (cfr. «Farmacocinetica»). Non si prevede un ritardo nel raggiungimento della massima attività.
Per gli eventi emorragici descritti di seguito, l'attività del fattore IX non deve scendere sotto il livello di attività plasmatica indicata (in % del normale o in UI/dl) nel periodo corrispondente. Le informazioni della Tabella 1 possono essere usate come guida al dosaggio in caso di episodi emorragici e interventi chirurgici.
Tabella 1: Guida al dosaggio di ALPROLIX per il trattamento di episodi emorragici e negli interventi chirurgici
Gravità dell'emorragia/tipo di intervento chirurgico | Livello di fattore IX richiesto (%) (UI/dl) | Frequenza di somministrazione (ore)/durata della terapia (giorni) |
Emorragia | ||
Emartrosi in fase iniziale, emorragia muscolare o emorragia orale | 20-40 | Ripetere l'iniezione ogni 48 ore fino a quando l'episodio emorragico - indicato dal dolore - è risolto o fino al raggiungimento della guarigione.1 |
Emartrosi più estesa, emorragia muscolare o ematoma | 30-60 | Ripetere l'iniezione ogni 24-48 ore fino alla risoluzione del dolore e dell'invalidità acuta.1 |
Emorragie pericolose per la vita | 60-100 | Ripetere l'iniezione ogni 8-24 ore finché il paziente non è fuori pericolo. |
Interventi chirurgici | ||
Interventi minori, estrazioni dentarie incluse | 30-60 | Ripetere l'iniezione ogni 24-48 ore, se necessario, fino a guarigione. |
Interventi chirurgici maggiori | 80-100 (pre- e post-operatorio) | Ripetere l'iniezione ogni 8-24 ore secondo necessità fino ad un'adeguata guarigione della ferità, quindi continuare la terapia per almeno 7 giorni per mantenere un'attività del fattore IX compresa tra il 30% e il 60% (UI/dl). |
1 Cfr. la Tabella 3 nella rubrica «Farmacocinetica».
Profilassi
Per la profilassi a lungo termine delle emorragie, si raccomandano i seguenti schemi posologici iniziali:
• 50 UI/kg una volta alla settimana, adeguando la dose in base alla risposta individuale del paziente o
• 100 UI/kg ogni 10 giorni, adeguando l'intervallo in base alla risposta individuale del paziente. Alcuni pazienti ben controllati con un regime di trattamento ogni 10 giorni possono eventualmente essere trattati con un intervallo di 14 giorni o più.
La dose massima raccomandata per la profilassi è di 100 UI/kg.
Istruzioni posologiche speciali
Pazienti anziani
Le esperienze nei pazienti di età ≥65 anni sono limitate.
Bambini e adolescenti
Nei bambini di età inferiore a 12 anni possono essere necessarie dosi più elevate o intervalli di dosaggio più brevi. La dose iniziale raccomandata è di 50-60 UI/kg ogni 7 giorni. Per gli adolescenti a partire dai 12 anni valgono le stesse raccomandazioni posologiche previste per gli adulti (cfr. «Proprietà/effetti» e «Farmacocinetica»). La dose massima raccomandata per la profilassi è di 100 UI/kg.
Sono disponibili dati limitati sul trattamento dei bambini di età inferiore a 6 anni durante gli interventi chirurgici.
Modo di somministrazione
Uso endovenoso.
ALPROLIX deve essere iniettato per via endovenosa nell'arco di diversi minuti. La velocità di somministrazione deve essere basata sulle condizioni del paziente e non deve superare i 10 ml/min.
L'uso come infusione continua non è omologato e non è raccomandato (cfr. anche «Avvertenze e misure precauzionali»).
Per le istruzioni sulla ricostituzione del medicamento prima dell'uso, cfr. «Indicazioni per la manipolazione».
Documentazione del numero di lotto
Per garantire la tracciabilità dei medicamenti biotecnologici, si raccomanda di prendere nota del nome commerciale e del numero di lotto in occasione di ogni trattamento.
Controindicazioni
Ipersensibilità al principio attivo o a una delle sostanze ausiliarie.
Avvertenze e misure precauzionali
Ipersensibilità
Durante la terapia con ALPROLIX sono state riportate reazioni da ipersensibilità di tipo allergico. I pazienti devono essere istruiti a interrompere immediatamente l'uso del medicamento e a contattare il medico se si verificano sintomi di una reazione da ipersensibilità. I pazienti devono essere informati dei primi segni di reazioni da ipersensibilità, quali orticaria, orticaria generalizzata, sensazione di costrizione toracica, respiro sibilante, ipotensione e anafilassi.
In caso di shock anafilattico deve essere istituita la terapia medica standard per il trattamento dello shock.
Inibitori
Dopo un trattamento ripetuto con preparati a base di fattore IX, i pazienti devono essere monitorati per lo sviluppo di anticorpi neutralizzanti (inibitori), da quantificare in unità Bethesda (UB) mediante appropriati test biologici.
I dati riportati in letteratura mostrano una correlazione tra la comparsa di un inibitore del fattore IX e le reazioni allergiche. Pertanto, nei pazienti che manifestano reazioni allergiche deve essere ricercata la presenza di inibitori. Inoltre, si deve tenere presente che i pazienti con inibitori del fattore IX possono presentare un rischio aumentato di anafilassi se il trattamento con il fattore IX viene proseguito.
A causa del rischio di reazioni allergiche dopo la somministrazione di preparati a base di fattore IX, le prime somministrazioni di fattore IX devono essere effettuate, secondo il giudizio del medico curante, sotto osservazione medica, in modo che, se necessario, siano disponibili le opzioni appropriate per il trattamento delle reazioni allergiche.
Tromboembolia
A causa del potenziale rischio di complicazioni tromboemboliche, l'uso di prodotti a base di fattore IX in pazienti con patologie epatiche, pazienti nel post-operatorio, neonati o pazienti a rischio di eventi trombotici o di coagulazione intravascolare disseminata (CID) deve essere monitorato clinicamente con adeguati test biologici per rilevare i segni precoci di una complicazione trombotica o di CID. In questi casi, il beneficio del trattamento con ALPROLIX deve essere valutato in rapporto al rischi di tali complicazioni.
Infusione continua
La sicurezza e l'efficacia dell'uso di ALPROLIX come infusione continua non sono dimostrate (cfr. anche «Posologia/impiego»).
Eventi cardiovascolari
In pazienti con fattori di rischio cardiovascolare esistenti, la terapia sostitutiva con prodotti a base di fattore IX può aumentare il rischio cardiovascolare.
Complicazioni associate a catetere
Se è necessario un catetere venoso centrale (CVC), si deve considerare il rischio di complicazioni associate a CVC, incluse infezioni locali, batteriemia e trombosi in sede di inserimento del catetere.
Bambini e adolescenti
Le «Avvertenze e misure precauzionali» elencate riguardano sia gli adulti sia i bambini.
Avvertenze sulle sostanze ausiliarie
Questo medicamento contiene meno di 1 mmol (23 mg) di sodio per flaconcino, cioè è essenzialmente «senza sodio». Contiene 0,3 mmol (o 6,4 mg) di sodio per flaconcino. Da tenere in considerazione soprattutto in pazienti che seguono una dieta a basso contenuto di sodio.
Interazioni
Non sono state riportate interazioni di ALPROLIX con altri medicamenti. Non sono stati effettuati studi di interazione.
Gravidanza/Allattamento
Non sono stati condotti studi sperimentali relativi alla funzione riproduttiva negli animali con ALPROLIX. Nel topo è stato effettuato uno studio di trasferimento placentare (cfr. «Dati preclinici»). Data la scarsa incidenza dell'emofilia B nelle donne, non sono disponibili esperienze sull'uso del fattore IX durante la gravidanza e l'allattamento. Pertanto, il fattore IX deve essere usato durante la gravidanza e l'allattamento solo se chiaramente indicato.
Effetti sulla capacità di condurre veicoli e sull'impiego di macchine
ALPROLIX non ha effetti sulla capacità di guidare veicoli o sulla capacità di utilizzare macchine.
Effetti indesiderati
Riassunto del profilo di sicurezza
In associazione con le terapie di sostituzione dei fattori, in rari casi sono possibili reazioni da ipersensibilità o reazioni allergiche, che possono includere angioedema, sensazione di bruciore e pizzicore in sede di infusione, brividi, sensazione di calore, orticaria generalizzata, orticaria, ipotensione, letargia, nausea, irrequietezza, tachicardia, sensazione di costrizione toracica, formicolio, vomito, respiro sibilante. In alcuni casi, queste reazioni sono progredite in anafilassi grave (incluso lo shock) e si sono verificate in stretta correlazione temporale con la formazione di inibitori del fattore IX (cfr. anche la rubrica «Avvertenze e misure precauzionali»). Nei pazienti con emofilia B si possono formare anticorpi neutralizzanti (inibitori) contro il fattore IX. In tal caso, lo sviluppo di inibitori si manifesta come risposta clinica insufficiente. Per questi casi si raccomanda di contattare un centro specializzato per la cura dell'emofilia. Sono stati riportati casi di sindrome nefrosica dopo il tentativo di induzione di tolleranza immunologica in pazienti affetti da emofilia B con inibitori del fattore IX e anamnesi di reazione allergica.
Esiste un rischio potenziale di episodi tromboembolici dopo somministrazione di preparati a base di fattore IX e tale rischio è maggiore per i prodotti a basso grado di purezza. L'uso di fattore IX ad alto grado di purezza è raramente associato a complicazioni tromboemboliche.
Tabella degli effetti collaterali
Pazienti precedentemente trattati (previously treated patient, PTP): la sicurezza di ALPROLIX è stata valutata in 153 pazienti con emofilia B grave in due studi di Fase 3 completati e in uno studio di estensione. Gli effetti indesiderati sono stati monitorati per un totale di 561 anni-paziente. Il numero totale di giorni di esposizione è stato di 26'106, con un numero mediano di 165 (range: 1-528) giorni di esposizione per paziente.
Pazienti precedentemente non trattati (previously untreated patient, PUP): un totale di 33 pazienti con emofilia B grave è stato osservato in uno studio clinico. Gli eventi indesiderati sono stati monitorati per un totale di 57,51 anni-paziente. Il numero totale di giorni di esposizione è stato di 2'233, con una mediana di 76 (range: 1-137) giorni di esposizione per paziente.
Gli effetti indesiderati più comunemente segnalati con ALPROLIX sono stati cefalea, parestesia orale, uropatia ostruttiva (PTP) ed eritema in sede di iniezione, con un'incidenza dell'1,3%, nonché inibizione del fattore IX e ipersensibilità nei PUP.
Gli effetti indesiderati sono elencati secondo la classificazione sistemica organica MedDRA (SOC e termini preferiti).
Le frequenze sono state valutate secondo la seguente convenzione: molto comune (≥1/10); comune (≥1/100, <1/10); non comune (≥1/1000, <1/100); raro (≥1/10'000, <1/1000); molto raro (<1/10000), singoli casi (la frequenza non può essere definita sulla base dei dati disponibili). La tabella elenca gli effetti collaterali che sono stati segnalati negli studi clinici o riscontrati durante la sorveglianza di post-marketing (dopo l'introduzione sul mercato).
Tabella 2: Effetti collaterali di ALPROLIX
Classe sistemica organica secondo MedDRA | Effetti collaterali | Categoria di frequenza |
Patologie del sistema emolinfopoietico | Inibizione del fattore IX | Comune1 |
Disturbi del sistema immunitario | Ipersensibilità Reazione anafilattica | Comune1 Singoli casi |
Disturbi del metabolismo e della nutrizione | Appetito ridotto | Non comune |
Patologie del sistema nervoso | Cefalea Capogiro Disturbo del gusto | Comune Non comune Non comune |
Patologie cardiache | Palpitazioni | Non comune |
Patologie vascolari | Ipotensione | Non comune |
Patologie gastrointestinali | Parestesia orale Alito cattivo | Comune Non comune |
Patologie renali e urinarie | Uropatia ostruttiva Ematuria Colica renale Sindrome nefrosica (dopo tentativo di induzione della tolleranza immunologica) | Comune Non comune Non comune Singoli casi |
Patologie generali e condizioni relative alla sede di somministrazione | Eritema in sede di iniezione Stanchezza Dolore in sede di infusione | Comune Non comune Non comune |
1 Frequenza basata sull'incidenza osservata nello studio sui PUP. Entrambi gli eventi di inibizione del fattore IX e ipersensibilità si sono verificati nello stesso PUP nello Studio IV. Cfr. «Descrizione di alcuni effetti collaterali».
Descrizione di alcuni effetti collaterali
Durante l'intero programma di studi clinici, un paziente (precedentemente non trattato) dello Studio IV ha sviluppato un basso titolo di inibitori del fattore IX associato a ipersensibilità (cfr. «Efficacia clinica»). Dopo l'introduzione sul mercato, sono stati osservati sviluppo di inibitori del fattore IX e ipersensibilità (inclusa anafilassi).
Bambini e adolescenti
Si prevede che la frequenza, il tipo e la severità degli effetti indesiderati nei bambini siano simili a quelli degli adulti. Per informazioni sull'entità e la struttura per età della banca dati di sicurezza nei bambini, cfr. «Farmacocinetica».
La notifica di effetti collaterali sospetti dopo l'omologazione del medicamento è molto importante. Consente una sorveglianza continua del rapporto rischio-beneficio del medicamento. Chi esercita una professione sanitaria è invitato a segnalare qualsiasi effetto indesiderato sospetto, nuovo o serio, attraverso il portale online ElViS (Electronic Vigilance System). Maggiori informazioni sul sito www.swissmedic.ch.
Posologia eccessiva
Non sono stati riportati casi di sovradosaggio.
Proprietà/Effetti
Codice ATC
B02BD04
Meccanismo d'azione
Eftrenonacog alfa (fattore IX della coagulazione umano ricombinante, proteina di fusione Fc (rFIXFc)) ha 867 aminoacidi. È prodotto utilizzando la tecnologia del DNA ricombinante in una linea di cellule embrionali renali umane (human embryonic kidney, HEK) e senza l'aggiunta di proteine esogene di origine umana o animale nel processo di coltura cellulare, nella purificazione o nella formulazione finale.
Il fattore IX è una glicoproteina a catena singola con una massa molecolare di circa 55'000 dalton. Questo fattore della coagulazione dipende dalla vitamina K. Il fattore IX è attivato dal fattore XIa nella via intrinseca della coagulazione e dal complesso fattore VII/fattore tissutale nella via estrinseca della coagulazione. In combinazione con il fattore VIII attivato, il fattore IX attivato attiva il fattore X. Il fattore X attivato converte la protrombina in trombina. La trombina converte quindi il fibrinogeno in fibrina, con conseguente formazione di un coagulo di sangue.
L'emofilia B è un disordine ereditario della coagulazione del sangue legato al sesso, causato da bassi livelli di fattore IX funzionale, che comporta emorragie nelle articolazioni, nei muscoli o negli organi interni, spontanee o secondarie a traumi accidentali o chirurgici. La terapia sostitutiva aumenta il livello di fattore IX nel plasma, consentendo di correggere temporaneamente il deficit di fattore IX e la tendenza al sanguinamento.
Farmacodinamica
ALPROLIX (eftrenonacog alfa) è una proteina di fusione interamente ricombinante a lunga durata d'azione, costituita dal fattore IX della coagulazione umano unito con legame covalente al dominio Fc dell'immunoglobulina G1 umana ed è prodotta mediante la tecnologia del DNA ricombinante.
La regione Fc dell'immunoglobulina G1 umana si lega al recettore neonatale dell'Fc. Questo recettore è espresso per tutta la vita e fa parte di una via metabolica naturale che protegge le immunoglobuline dalla degradazione lisosomiale, riportando queste proteine nel torrente ematico, il che ne determina la lunga emivita plasmatica.
Efficacia clinica
La sicurezza, l'efficacia e la farmacocinetica di ALPROLIX sono state valutate in due studi cardine multinazionali, in aperto, su pazienti precedentemente trattati (PTP): uno studio di Fase 3 (Studio I in adulti e adolescenti) e uno studio pediatrico di Fase 3 (Studio II) (cfr. «Bambini e adolescenti»). La sicurezza e l'efficacia di ALPROLIX sono state valutate anche in pazienti precedentemente non trattati (PUP) affetti da emofilia B grave (Studio IV); cfr. «Bambini e adolescenti».
Lo Studio I ha confrontato l'efficacia di due regimi di trattamento profilattico (intervallo settimanale fisso con una dose di 50 UI/kg e intervallo di dosaggio personalizzato con 100 UI/kg ogni 10 giorni) con quella del trattamento al bisogno. Nello studio è stato arruolato un totale di 123 pazienti di sesso maschile precedentemente trattati (di età compresa tra 12 e 71 anni) affetti da emofilia B grave (attività endogena di FIX ≤2%). Tutti i pazienti hanno ricevuto ALPROLIX e sono stati seguiti per un massimo di 77 settimane.
Dei 123 pazienti che hanno completato lo Studio I, 93 sono stati arruolati nello studio di estensione (Studio III), con un periodo di follow-up di 6,5 anni.
Si noti che i tassi annualizzati di sanguinamento (Annualised Bleeding Rate, ABR) di differenti concentrati di fattori della coagulazione e differenti studi clinici non sono paragonabili.
Profilassi con intervallo di dosaggio settimanale fisso e personalizzato:
La dose settimanale mediana per i pazienti nel braccio con intervallo di dosaggio settimanale fisso è stata di 45,17 UI/kg (intervallo interquartile [interquartile range, IQR]: 38,1-53,7) nello Studio I. I corrispondenti ABR mediani nei pazienti valutabili ai fini dell'efficacia sono stati di 2,95 (IQR: 1,01-4,35) e sono rimasti simili durante lo Studio III (1,85 [IQR: 0,76-4,0]). I pazienti nello Studio III presentavano una mediana di 0,38 (IQR: 0,00-1,43) emorragie articolari spontanee.
Per i pazienti nel braccio con intervallo di dosaggio personalizzato nello Studio I, l'intervallo di dosaggio mediano è stato pari a 12,53 giorni (IQR: 10,4-13,4). I corrispondenti ABR mediani sono stati pari a 1,38 (IQR: 0,00-3,43) e sono rimasti simili durante lo Studio III (1,85 [IQR: 0,76-4,0]).
Gli intervalli di dosaggio e il consumo del fattore sono stati simili nello Studio III (studio di estensione) e nello Studio I per entrambi i regimi di trattamento in profilassi.
Nessun episodio emorragico si è verificato nel 42% dei pazienti in regime di profilassi personalizzata e nel 23,0% dei pazienti in regime di profilassi settimanale. Nei pazienti in regime di profilassi con intervallo di dosaggio personalizzato è stata osservata una percentuale minore di soggetti con ≥1 articolazione target al basale rispetto ai soggetti in regime di profilassi settimanale (rispettivamente 27,6% e 57,1%).
Trattamento delle emorragie:
Dei 636 eventi emorragici osservati nello Studio I, il 90,4% è stato controllato con 1 iniezione e complessivamente il 97,3% è stato controllato con 2 iniezioni o meno. La dose mediana per iniezione per il trattamento di un episodio emorragico è stata di 46,07 UI/kg (IQR: 32,86-57,03). La dose mediana complessiva per il trattamento di un episodio emorragico è stata di 51,47 UI/kg (IQR 35,21-61,73) nel braccio di profilassi settimanale, 49,62 UI/kg (IQR: 35,71-94,82) nel braccio di profilassi con intervallo personalizzato e 46,58 UI/kg (IQR: 33,33-59,41) nel braccio con trattamento al bisogno.
Gestione perioperatoria (profilassi chirurgica):
Un totale di 35 interventi di chirurgia maggiore è stato effettuato e valutato in 22 pazienti (21 adulti e adolescenti e 1 bambino <12 anni) nello Studio I e nello Studio III. Dei 35 interventi di chirurgia maggiore, 28 interventi (80,0%) hanno richiesto la somministrazione di una singola dose preoperatoria per il mantenimento dell'emostasi durante l'intervento. La mediana della dose media per iniezione per il mantenimento dell'emostasi durante l'intervento è stata pari a 94,7 UI/kg (range: 49-152 UI/kg). La dose totale il giorno dell'intervento era compresa tra 49 e 341 UI/kg e la dose totale nel periodo perioperatorio di 14 giorni era compresa tra 60 e 1'947 UI/kg.
La risposta emostatica è stata giudicata eccellente o buona nel 100% degli interventi di chirurgia maggiore.
Bambini e adolescenti
Nello Studio II è stato arruolato un totale di 30 pazienti pediatrici di sesso maschile precedentemente trattati affetti da emofilia B grave (attività endogena di FIX ≤2%). I pazienti avevano un'età inferiore a 12 anni (15 avevano <6 anni e 15 erano di età compresa tra 6 e <12 anni). Tutti i pazienti hanno ricevuto ALPROLIX e sono stati seguiti per un massimo di 52 settimane.
Tutti e 30 i pazienti sono stati trattati con ALPROLIX nel quadro di un regime di trattamento in profilassi a una dose iniziale di 50-60 UI/kg ogni 7 giorni, con adeguamenti della dose fino a un massimo di 100 UI/kg e un intervallo di dosaggio da un minimo di una a un massimo di due volte a settimana.
Dei 30 pazienti che hanno completato lo Studio II, 27 sono stati arruolati nello Studio III (studio di estensione). Il tempo mediano di permanenza negli Studi II+III è stato pari a 2,88 anni e il numero mediano di giorni di esposizione è stato pari a 166.
Nello Studio IV sono stati arruolati 33 pazienti pediatrici precedentemente non trattati (PUP) affetti da emofilia B grave (attività endogena di FIX ≤2%). L'età mediana all'arruolamento era 0,6 anni (range: 0,08-2 anni); il 78,8% dei partecipanti aveva meno di 1 anno di età. La durata mediana complessiva del trattamento con ALPROLIX è stata di 83,01 settimane (range: 6,7-226,7 settimane), con un numero mediano complessivo di 76 giorni (range: 1-137 giorni).
Profilassi con regime di trattamento personalizzato:
Nello Studio II, la mediana della dose settimanale media di ALPROLIX è stata di 59,40 UI/kg (intervallo interquartile: 52,95-64,78 UI/kg) per i pazienti di età <6 anni e 57,78 UI/kg (intervallo interquartile: 51,67-65,01 UI/kg) per i pazienti di età compresa tra 6 e <12 anni. Complessivamente, l'intervallo di dosaggio mediano è stato pari a 6,99 giorni (intervallo interquartile: 6,94-7,03). Non vi sono state differenze tra i due gruppi di età per quanto riguarda gli intervalli di dosaggio mediani. Ad eccezione di un paziente per il quale l'ultima dose prescritta è stata di 100 UI/kg ogni 5 giorni, negli altri 29 pazienti le ultime dosi prescritte ammontavano ad un massimo di 70 UI/kg ogni 7 giorni. Il 33% dei pazienti pediatrici non ha manifestato alcun episodio emorragico. Gli intervalli di dosaggio e il consumo del fattore sono stati simili fra lo Studio III e lo Studio II.
I tassi annualizzati mediani di sanguinamento nei pazienti di età <12 anni valutabili ai fini dell'efficacia erano di 1,97 (intervallo interquartile: 0,00-3,13) nello Studio II e sono rimasti simili nel corso dello Studio III (studio di estensione).
Nei PUP (Studio IV), la mediana della dose settimanale media di ALPROLIX è stata di 57,96 UI/kg (intervallo interquartile: 52,45-65,06 UI/kg) e l'intervallo di dosaggio mediano è stato pari a 7 giorni (intervallo interquartile: 6,95-7,12 giorni). Gli intervalli di dosaggio e il consumo del fattore sono stati simili fra Studio IV e gli Studi II e III. Tra i PUP sottoposti a profilassi, 8 (28,6%) soggetti non hanno manifestato episodi emorragici. L'ABR mediano tra tutti i soggetti in regime di profilassi è stato di 1,24 (intervallo interquartile: 0,0-2,49).
Trattamento degli episodi emorragici:
Dei 60 eventi emorragici osservati nello Studio II, il 75% è stato controllato con 1 iniezione e complessivamente il 91,7% è stato controllato con 2 iniezioni o meno. La dose mediana per iniezione per il trattamento di un episodio emorragico è stata di 63,51 UI/kg (intervallo interquartile: 48,92-99,44). La dose mediana complessiva per il trattamento di un episodio emorragico è stata di 68,22 UI/kg (intervallo interquartile: 50,89-126,19).
Dei 58 eventi emorragici osservati in PUP sottoposti a profilassi nello Studio IV, l'87,9% è stato controllato con 1 iniezione e complessivamente il 96,6% è stato controllato con 2 iniezioni o meno. La mediana della dose media per iniezione per il trattamento di un episodio emorragico è stata di 71,92 UI/kg (intervallo interquartile: 52,45-100,81 UI/kg). La dose mediana complessiva per il trattamento di un episodio emorragico è stata di 78,74 UI/kg (intervallo interquartile: 53,57-104,90 UI/kg).
Farmacocinetica
Assorbimento
Tutti gli studi farmacocinetici con ALPROLIX sono stati condotti in pazienti con emofilia B grave precedentemente trattati. I dati presentati in questa rubrica sono stati ottenuti mediante test di coagulazione one-stage con un reagente aPTT a base di silice calibrato con uno standard plasmatico di fattore IX. Le proprietà farmacocinetiche sono state valutate in 22 pazienti (≥19 anni) trattati con ALPROLIX (rFIXFc). I campioni per gli studi di farmacocinetica sono stati raccolti prima della somministrazione di 50 UI/kg di rFIX (Benefix) e in 8 punti temporali in un periodo fino a 96 ore (4 giorni) dopo la somministrazione. Dopo un periodo di washout di almeno 120 ore (5 giorni), i pazienti hanno ricevuto una singola dose di ALPROLIX da 50 UI/kg. I campioni per gli studi di farmacocinetica sono stati raccolti prima della somministrazione e, successivamente, in 11 punti temporali in un periodo fino a 240 ore (10 giorni) dopo la somministrazione. I parametri farmacocinetici dopo somministrazione di una dose da 50 UI/kg di ALPROLIX sono riportati nella Tabella 3.
Tabella 3: Parametri farmacocinetici di ALPROLIX (50 UI/kg)
Parametri farmacocinetici1 | ALPROLIX |
N= 22 | |
Recupero incrementale (UI/dl per UI/kg) | 0,92 |
AUC/dose (UI*h/dl per UI/kg) | 31,32 |
Cmax (UI/dl) | 40,81 |
Cl (ml/h/kg) | 3,19 |
t1/2α (h) | 5,03 |
t1/2β (h) | 82,12 |
MRT (h) | 98,60 |
Vss (ml/kg) | 314,80 |
Tempo al 1% (giorni) | 11,22 (10,20-12,35) |
1 I parametri farmacocinetici sono presentati come media geometrica (IC 95%)
Abbreviazioni: IC = intervallo di confidenza; Cmax = attività massima; AUC = area sotto la curva
(area under the curve) di attività del FIX/tempo; t1/2α = emivita di distribuzione;
t1/2β = emivita di eliminazione; Cl = clearance; Vss = volume di distribuzione allo stato stazionario;
MRT = tempo medio di permanenza (mean residence time).
L'emivita di eliminazione (82 ore) è influenzata dalla regione Fc, che in modelli animali si è dimostrata mediata dalle vie metaboliche del recettore Fc neonatale.
È stato sviluppato un modello farmacocinetico di popolazione basato sui dati dell'attività del FIX di 161 pazienti di tutte le età (2-76 anni) con peso compreso tra 12,5 kg e 186,7 kg partecipanti a tre studi clinici (12 pazienti di uno studio di Fase 1/2a, 123 pazienti dello Studio I e 26 pazienti dello Studio II). La clearance di ALPROLIX attesa in un adulto tipico di 70 kg è di 2,30 dl/h e il volume di distribuzione allo stato stazionario di ALPROLIX è di 194,8 dl. Il modello è stato utilizzato per prevedere il profilo di attività/tempo dopo una singola dose di ALPROLIX in pazienti con emofilia B grave (cfr. Tabella 4).
Tabella 4: Attività di FIX prevista [UI/dl] dopo una singola dose di ALPROLIX1 per pazienti di età ≥12 anni
Dose (UI/kg) | Fine dell'iniezione | 12 ore | 24 ore (Giorno 1) | 36 ore | 48 ore (Giorno 2) | 72 ore (Giorno 3) | Giorno 5 | Giorno 7 | Giorno 10 | Giorno 14 |
Mediana (5., 95.) | ||||||||||
50 | 52,0 | 21,7 | 15,2 | 11,3 | 8,4 | 5,5 | 3,02 | 1,93 | 1,07 | 0,495 |
[31,8; 82] | [15,0; 31,0] | [10,2; 21,5] | [7,50; 16,5] | [5,5; 12,5] | [3,59; 8,25] | [1,88; 4,65] | [0,99; 3,12] | [0,345; 1,95] | [0,0829; 1,17] | |
100 | 104,0 | 43,4 | 30,4 | 22,6 | 16,8 | 11 | 6,03 | 3,85 | 2,13 | 0,991 |
[63,6; 164] | [30,0; 62,0] | [20,4; 42,9] | [15,0; 32,9] | [11; 24,9] | [7,18; 16,5] | [3,77; 9,29 | [1,98; 6,24] | [0,691; 3,89] | [0,166; 2,34] | |
1 Cfr. «Posologia/impiego»
Distribuzione
Cfr. «Assorbimento».
Metabolismo
Cfr. «Assorbimento».
Eliminazione
Cfr. «Assorbimento».
Cinetica di gruppi di pazienti speciali
Bambini e adolescenti
I parametri farmacocinetici di ALPROLIX sono stati determinati negli adolescenti nello Studio I e nei bambini nello Studio II (i campioni farmacocinetici sono stati raccolti prima della somministrazione e in più punti temporali in un periodo fino a 336 ore (14 giorni) dopo la somministrazione nello Studio I e prima della somministrazione e in 7 punti temporali in un periodo fino a 168 ore (7 giorni) dopo la somministrazione nello Studio II, rispettivamente).
La Tabella 5 riporta i parametri farmacocinetici calcolati a partire dai dati pediatrici di 35 pazienti di età inferiore a 18 anni.
Tabella 5: Confronto dei parametri farmacocinetici di ALPROLIX (rFIXFc) per fascia d'età
Parametri farmacocinetici1 | Studio II | Studio I | |
<6 anni (range: 2-4) | Da 6 a <12 anni (range: 6-10) | Da 12 a <18 anni (range: 12-17) | |
N = 11 | N = 13 | N = 11 | |
RI (UI/dl per UI/kg) | 0,5898 | 0,7170 | 0,8470 |
AUC/dose (UI*h/dl per UI/kg) | 22,71 | 28,53 | 29,50 |
t½ (h) | 66,49 | 70,34 | 82,22 |
MRT (h) | 83,65 | 82,46 | 93,46 |
Cl (ml/h/kg) | 4,365 | 3,505 | 3,390 |
Vss (ml/kg) | 365,1 | 289,0 | 316,8 |
1 I parametri farmacocinetici ottenuti nell'analisi non compartimentale sono presentati come media geometrica (IC 95%) Abbreviazioni: IC = intervallo di confidenza; RI = recupero incrementale; AUC = area sotto la curva (area under the curve) di attività del FIX/tempo; t1/2= emivita terminale; MRT = tempo medio di permanenza (mean residence time); Cl = clearance; Vss = volume di distribuzione allo stato stazionario | |||
Dati preclinici
I dati preclinici degli studi su tossicità acuta e tossicità per somministrazione ripetuta, che hanno incluso valutazioni di tossicità locale e farmacologia di sicurezza, non evidenziano alcun rischio particolare per l'essere umano. Non sono stati effettuati studi di genotossicità, cancerogenicità, tossicità riproduttiva o dello sviluppo embrionale/fetale. In uno studio di trasferimento placentare è stato evidenziato che, nel topo, eftrenonacog alfa (rFIXFc) attraversa la placenta in piccole concentrazioni.
Fertilità
Non sono stati effettuati studi sulla fertilità animale con ALPROLIX.
Altre indicazioni
Incompatibilità
Poiché non sono stati condotti studi di compatibilità, non si può somministrare questo medicamento in combinazione con altri medicamenti.
Utilizzare solo il set di somministrazione fornito in dotazione, poiché il trattamento può essere reso inefficace da un adsorbimento del fattore IX della coagulazione alla superficie interna di alcuni dispositivi per infusione.
Stabilità
Il medicamento non deve essere utilizzato oltre la data indicata con «EXP» sul contenitore. Nel periodo di stabilità, il medicamento può essere conservato a temperatura ambiente (fino a 30 °C) per un singolo periodo non superiore a 6 mesi. La data di prelevamento del medicamento dal frigorifero deve essere annotata sulla scatola. Dopo essere stato conservato a temperatura ambiente, il medicamento non deve essere posto nuovamente in frigorifero.
Dopo la ricostituzione
La stabilità chimico-fisica è stata dimostrata per 6 ore a temperatura ambiente (fino a 30 °C). Il medicamento non utilizzato entro 6 ore deve essere smaltito. Per ragioni microbiologiche, il medicamento dovrebbe essere utilizzato immediatamente dopo la ricostituzione. Qualora ciò non sia possibile, i tempi e le condizioni di conservazione sono di responsabilità dell'utilizzatore. Proteggere il medicamento dalla luce solare diretta.
Precauzioni particolari per la conservazione
Tenere il medicamento fuori dalla portata dei bambini.
Conservare il flaconcino nella scatola originale per proteggere il contenuto dalla luce. Conservare in frigorifero (2-8 °C). Non congelare. Questo medicamento è monouso. La soluzione residua deve essere smaltita correttamente.
Per le condizioni di conservazione dopo la ricostituzione del medicamento, cfr. «Stabilità».
Indicazioni per la manipolazione
La polvere liofilizzata per soluzione iniettabile contenuta nel flaconcino deve essere ricostituita con il solvente (soluzione di sodio cloruro) fornito nella siringa preriempita utilizzando l'adattatore sterile per flaconcino. Il flaconcino deve essere ruotato leggermente fino a completa dissoluzione della polvere.
La soluzione ricostituita deve essere da limpida a leggermente opalescente e incolore. Prima della somministrazione, il medicamento ricostituito deve essere ispezionato visivamente per l'eventuale presenza di particelle e alterazioni del colore. Non utilizzare soluzioni torbide o con depositi.
Questo prodotto è esclusivamente monouso. ALPROLIX non deve essere miscelato con altre soluzioni iniettabili o per infusione.
Il medicamento non utilizzato e i rifiuti derivati da tale medicamento devono essere smaltiti correttamente.
Una confezione di ALPROLIX contiene:
| A) 1 flaconcino con polvere |

Preparazione:
Lavarsi le mani prima di aprire la confezione.
1. Controllare la denominazione e il dosaggio riportati sulla confezione per assicurarsi che questa contenga il prodotto corretto. Controllare la data di scadenza sulla scatola di ALPROLIX. Non usare se il medicamento è scaduto. | |
2. Se ALPROLIX è stato conservato in frigorifero, attendere che il flaconcino di ALPROLIX (A) e la siringa con il solvente (B) raggiungano la temperatura ambiente prima dell'uso. Non usare fonti di calore esterne. | |
3. Appoggiare il flaconcino su una superficie piana pulita. Rimuovere la capsula di chiusura in plastica dal flaconcino. |
|
4. Pulire la parte superiore del flaconcino con uno dei tamponi imbevuti di alcol (F) inclusi nella confezione e lasciarla asciugare all'aria. Una volta che è stata pulita, non toccare in alcun modo la parte superiore del flaconcino e non farla entrare in contatto con altri oggetti. |
|
5. Rimuovere la carta protettiva dall'adattatore per flaconcino in plastica trasparente (D). Non togliere l'adattatore dalla sua capsula di chiusura protettiva. Non toccare l'interno della confezione dell'adattatore per flaconcino. | |
6.Appoggiare il flaconcino su una superficie piana. Afferrare saldamente l'adattatore per flaconcino per la capsula di chiusura protettiva e posizionarlo perpendicolarmente sopra il flaconcino. Premere con decisione verso il basso l'adattatore fino a bloccarlo sulla parte superiore del flaconcino, con la punta che perfora il tappo del flaconcino. |
|
7.Collegare lo stantuffo (C) alla siringa del solvente inserendo la punta dello stantuffo nel cilindro della siringa. Ruotare con decisione lo stantuffo in senso orario fino a fissarlo saldamente nella siringa. |
|
8. Rompere la capsula di chiusura antimanomissione in plastica bianca della siringa del solvente piegandola verso il basso, dove perforata, fino a romperla. Appoggiare la capsula di chiusura su una superficie piana con la parte superiore rivolta verso il basso. Non toccare l'interno della capsula di chiusura o la punta della siringa. |
|
9. Rimuovere la capsula di chiusura protettiva dall'adattatore ed eliminarla. |
|
10. Collegare la siringa del solvente all'adattatore per flaconcino inserendo la punta della siringa nell'apertura dell'adattatore. Premere con decisione e ruotare la siringa in senso orario fino a fissarla saldamente. |
|
11. Premere lentamente lo stantuffo verso il basso per iniettare tutto il solvente nel flaconcino di ALPROLIX. |
|
12. Con la siringa ancora collegata all'adattatore e continuando a premere lo stantuffo, ruotare leggermente il flaconcino fino alla dissoluzione della polvere. Non agitare. |
|
13. La soluzione finale deve essere ispezionata visivamente prima della somministrazione. La soluzione deve essere da limpida a leggermente opalescente e incolore. Non utilizzare soluzioni torbide o con particelle visibili. | |
14. Assicurarsi che lo stantuffo nella siringa sia ancora completamente premuto, quindi capovolgere il flaconcino. Tirare indietro lentamente lo stantuffo per aspirare tutta la soluzione nella siringa attraverso l'adattatore per flaconcino. Nota: se si usa più di un flaconcino di ALPROLIX per iniezione, ogni flaconcino deve essere preparato separatamente seguendo le precedenti istruzioni (punti 1-13). Rimuovere la siringa del solvente lasciando collegato l'adattatore per flaconcino. Per aspirare le soluzioni ricostituite dai singoli flaconcini, si può utilizzare un'unica siringa grande con attacco Luer Lock. |
|
15. Staccare la siringa dall'adattatore per flaconcino tirando delicatamente il flaconcino e ruotandolo in senso antiorario. |
|
16. Eliminare il flaconcino e l'adattatore. Nota: se la soluzione non viene utilizzata immediatamente, il cappuccio della siringa deve essere riposto sulla siringa. Non toccare la punta della siringa o l'interno della capsula di chiusura. Dopo la preparazione, ALPROLIX può essere conservato a temperatura ambiente per un massimo di 6 ore prima della somministrazione. Una volta trascorso questo tempo, ALPROLIX deve essere eliminato. Proteggere dalla luce solare diretta. | |
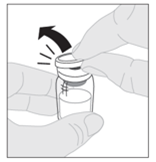










Somministrazione:
1.Aprire la confezione del set per infusione e rimuovere la capsula di chiusura all'estremità del tubicino. Collegare la siringa contenente la soluzione preparata di ALPROLIX all'estremità del tubicino del set per infusione ruotando in senso orario. |
|
2.Se necessario, applicare un laccio emostatico e preparare la sede di iniezione strofinando bene la cute con il secondo tampone imbevuto di alcol incluso nella confezione.
| |
3. Rimuovere tutta l'aria eventualmente presente nel tubicino del set per infusione premendo lentamente lo stantuffo fino a che il liquido non abbia raggiunto l'ago del set per infusione. Non spingere la soluzione nell'ago. Togliere dall'ago il cappuccio protettivo in plastica trasparente. | |
4. Inserire l'ago del set per infusione in una vena, secondo le istruzioni ricevute dal medico o dal personale sanitario, e rimuovere il laccio emostatico. Se lo si desidera, si può usare uno dei cerotti (G) inclusi nella confezione per fissare le alette in plastica dell'ago nella sede di iniezione. Il medicamento preparato deve essere iniettato per via endovenosa nell'arco di diversi minuti. Per un trattamento più confortevole, il medico può modificare la velocità di iniezione raccomandata. | |
5. Dopo aver completato l'iniezione e aver rimosso l'ago, coprire l'ago con il cappuccio protettivo, chiudendolo fino a bloccarlo. |
|
6.Smaltire in sicurezza l'ago usato, la soluzione non utilizzata, la siringa e il flaconcino vuoto in un apposito contenitore per rifiuti sanitari, poiché questi materiali possono arrecare danni a terzi se non vengono smaltiti correttamente. Non riutilizzare il materiale usato. | |



Numero dell'omologazione
66039 (Swissmedic)
Titolare dell’omologazione
Swedish Orphan Biovitrum AG, Basel
Stato dell'informazione
Dicembre 2021
Composition
Principes actifs
eftrenonacog alfa (facteur de coagulation IX humain (ADNr))
Excipients
Poudre: saccharose, histidine, mannitol, polysorbate 20, hydroxyde de sodium (pour l'ajustement du pH), acide chlorhydrique (pour l'ajustement du pH)
Solvants: solution de chlorure de sodium, eau pour injection
Sodium par flacon: 0,3 mmol (6,4 mg).
Forme pharmaceutique et quantité de principe actif par unité
Poudre et solvant pour solution injectable (i.v.)
1 flacon de poudre ALPROLIX contient de l'eftrenonacog alfa dans les concentrations suivantes:
250 U.I d'eftrenonacog alfa (50 U.I./ml après reconstitution avec 5 ml de solvant (solution de chlorure de sodium))
500 U.I d'eftrenonacog alfa (100 U.I./ml après reconstitution avec 5 ml de solvant (solution de chlorure de sodium))
1000 U.I d'eftrenonacog alfa (200 U.I. /ml après reconstitution avec 5 ml de solvant (solution de chlorure de sodium))
2000 U.I d'eftrenonacog alfa (400 U.I./ml après reconstitution avec 5 ml de solvant (solution de chlorure de sodium))
3000 U.I d'eftrenonacog alfa (600 U.I./ml après reconstitution avec 5 ml de solvant (solution de chlorure de sodium))
Indications/Possibilités d’emploi
ALPROLIX est indiqué pour le traitement et le traitement préventif des saignements chez les patients atteints d'hémophilie B (déficit congénital en facteur IX) et déjà traités auparavant.
Posologie/Mode d’emploi
Le traitement doit se faire sous le contrôle d'un médecin expérimenté dans le traitement de l'hémophilie.
Surveillance de la thérapie
Pendant la thérapie, une détermination adéquate du taux de facteur IX est conseillée afin d'adapter au mieux la posologie et la fréquence des injections répétées à effectuer. La réponse de chaque patient au facteur IX peut varier, ce qui se manifeste par des demi-vies différentes et des récupérations différentes. En ce qui concerne la posologie basée sur le poids corporel, il peut s'avérer nécessaire de procéder à un ajustement chez les patients en surcharge ou en insuffisance pondérale. C'est surtout lors d'interventions chirurgicales importantes qu'une surveillance précise de la thérapie de substitution à l'aide d'une analyse de la coagulation (activité du facteur IX dans le plasma) est indispensable.
Lors de l'utilisation d'un test de coagulation in-vitro en une seule étape sur la base du temps de céphaline activée (aPTT) afin de déterminer l'activité du facteur IX dans les échantillons sanguins du patient, les résultats de l'activité du facteur IX peuvent être considérablement influencés aussi bien par le type de réactif aPTT utilisé que par le témoin de référence utilisé. Ce point doit être tout particulièrement pris en compte en cas de changement de laboratoire et/ou des réactifs utilisés dans le test.
Les mesures effectuées à l'aide d'un test de coagulation en une seule étape au cours duquel un réactif aPTT à base de kaoline est utilisé conduisent probablement à une sous-évaluation du taux d'activité.
Posologie
La posologie et la durée de la thérapie de substitution se basent sur le degré de gravité du déficit en facteur IX, la localisation et l'étendue du saignement, ainsi que l'état clinique du patient.
Le nombre d'unités du facteur IX à appliquer est indiqué en Unités Internationales (U.I.) sur la base de la norme actuelle de l'OMS pour les préparations de facteur IX. L'activité du facteur IX dans le plasma est représentée en pourcentage (par rapport à un plasma humain normal) ou en unités internationales (par rapport à une norme internationale pour le facteur IX dans le plasma).
Une unité internationale (U.I.) de facteur IX Fc recombinant correspond à la quantité de facteur IX dans un ml de plasma humain normal.
Traitement en fonction de la situation
Le calcul de la dose nécessaire de facteur IX Fc recombinant se base sur le résultat empirique selon lequel 1 unité internationale (U.I.) de facteur IX par kg de poids corporel augmente l'activité du facteur IX dans le plasma de 1 % par rapport à l'activité normale (U.I./dl). La dose nécessaire se calcule avec la formule suivante:
Unités nécessaires = poids corporel (kg) x augmentation souhaitée du facteur -IX (%) (U.I.) x valeur inverse de la récupération déterminée (U.I./kg par U.I./dl).
La dose nécessaire, ainsi que la fréquence de l'application, doivent toujours s'orienter par rapport à l'efficacité clinique au cas par cas. Si le contrôle des saignements nécessite une administration supplémentaire, il convient alors de tenir compte de la durée de demi-vie d'ALPROLIX la plus longue (voir «pharmacocinétique»). Un retard de la durée jusqu'à l'atteinte de l'activité maximale n'est pas à attendre.
Lors des saignements décrits ci-après, l'activité du facteur IX ne doit pas descendre en dessous de l'activité plasmatique indiquée (en % de la norme, ou en U.I./dl) sur la période correspondante. Les données figurant dans le tableau 1 peuvent servir de valeurs indicatives pour la posologie lors des épisodes hémorragiques et les interventions chirurgicales.
Tableau 1: directives pour la posologie d'ALPROLIX dans le cadre du traitement des épisodes hémorragiques et lors d'interventions chirurgicales
Degré de gravité du saignement / Nature de l'intervention chirurgicale | Taux de facteur IX nécessaire (%) (U.I./dl) | Fréquence de l'application (en heures) / durée de la thérapie (en jours) |
Saignement | ||
Hémarthrose à un stade précoce, saignement musculaire ou saignement buccal | 20-40 | Répéter l'injection toutes les 48 heures, jusqu'à ce que l'épisode hémorragique (identifié par les douleurs) se soit atténué ou qu'une guérison ait été atteinte.1 |
Hémarthrose étendue, saignement musculaire ou hématome | 30-60 | Répéter l'injection toutes les 24 à 48 heures, jusqu'à ce que les douleurs et les troubles aigus aient disparu.1 |
Saignements dangereux pour la vie du patient | 60-100 | Répéter l'injection toutes les 8 à 24 heures, jusqu'à ce que le pronostic vital du patient ne soit plus engagé. |
Interventions chirurgicales | ||
Interventions de petite envergure, y compris extraction dentaire | 30-60 | Si nécessaire, répéter l'injection toutes les 24 à 48 heures, jusqu'à ce qu'une guérison ait été atteinte. |
Interventions chirurgicales de grande envergure | 80-100 (pré- et postopératoires) | Si nécessaire, répéter l'injection toutes les 8 à 24 heures, jusqu'à ce que la plaie ait suffisamment cicatrisé, puis poursuivre la thérapie pendant au moins 7 jours, afin de maintenir une activité du facteur IX de 30% à 60% (U.I./dl). |
1 Voir le tableau 3 au paragraphe «Pharmacocinétique».
Traitement préventif
Pour la prévention de longue durée des saignements, sont recommandées les posologies initiales suivantes:
• 50 U.I./kg une fois par semaine, ajustement de la posologie selon la réponse individuelle du patient, ou
• 100 U.I./kg tous les 10 jours, ajustement de l'intervalle selon la réponse individuelle du patient. Chez certains patients, lorsque les saignements sont bien contrôlés par le traitement administré une fois tous les 10 jours, un allongement de l'intervalle de traitement à 14 jours ou plus peut être possible.
La posologie maximale conseillée pour le traitement préventif est de 100 U.I./kg.
Instructions spéciales pour la posologie
Patients âgés
L'expérience avec les patients âgés de 65 ans et plus est limitée.
Enfants et adolescents
Chez les enfants de moins de 12 ans, il peut être nécessaire d'appliquer des doses plus élevées, ou des intervalles d'administration plus courts. La posologie initiale recommandée est de 50-60 U.I./kg tous les 7 jours. Pour les adolescents à partir de 12 ans, les mêmes recommandations de posologie que pour les adultes sont applicables (voir «Propriétés/Effets» et «Pharmacocinétique»). La dose maximale recommandée pour le traitement préventif est de 100 U.I./kg.
Les données disponibles pour le traitement des enfants de moins de 6 ans lors d'interventions chirurgicales sont limitées.
Type d'application
Application intraveineuse
ALPROLIX doit être injecté par voie intraveineuse sur une durée de plusieurs minutes. La vitesse d'administration être déterminée en fonction de l'état du patient et ne doit pas dépasser 10 ml/min.
Une utilisation sous forme de perfusion continue n'est pas autorisée, elle n'est donc pas recommandée (voir «Mises en garde et précautions»).
Pour les remarques sur la reconstitution du médicament avant l'utilisation, voir «Remarques concernant la manipulation».
Documentation du numéro de lot
Afin d'assurer la traçabilité des médicaments fabriqués à l'aide de la biotechnologie, il est conseillé de documenter le nom commercial et le numéro de lot pour chaque traitement.
Contre-indications
Hypersensibilité au principe actif ou à l'un des excipients.
Mises en garde et précautions
Hypersensibilité
Lors d'une thérapie à base d'ALPROLIX, des réactions d'hypersensibilité allergique ont été rapportées. Il convient d'ordonner aux patients de stopper immédiatement l'utilisation du médicament et de s'adresser à leur médecin si des symptômes d'hypersensibilité apparaissent. Les patients doivent être informés des premiers signes des réactions d'hypersensibilité, y compris papules, urticaire généralisé, sensations d'oppression dans la poitrine, sifflements, hypotonie et chocs anaphylactiques.
Dans le cas d'un choc anaphylactique, il convient d'appliquer la thérapie médicale standard pour le traitement du choc.
Inhibiteurs
Après un traitement répété par des préparations de facteur IX, les patients doivent être surveillés afin de détecter l'éventuel développement d'anticorps neutralisants (inhibiteurs). Ces derniers doivent être quantifiés au moyen de tests biologiques adaptés et exprimés en unités Bethesda (U.B.).
Les rapports publiés dans la littérature spécialisée montrent une corrélation entre l'apparition d'un inhibiteur du facteur IX et les réactions allergiques. C'est pourquoi les patients présentant des réactions allergiques doivent être testés afin de détecter la présence d'éventuels inhibiteurs. Il convient de noter que les patients ayant des inhibiteurs de facteur IX présentent un risque plus important de réactions anaphylactiques en cas d'exposition ultérieure au facteur IX.
En raison du risque de réactions allergiques après l'administration de préparations de facteur IX, il convient de réaliser les premières applications de facteur IX selon l'estimation du médecin traitant et sous observation médicale dans des établissements équipés pour traiter les réactions allergiques.
Thromboembolie
En raison du risque potentiel de complications thromboemboliques, l'utilisation de produits à base de facteur IX chez les patients souffrant d'une maladie du foie, les patients postopératoires, les nouveau-nés ou les patients présentant un risque d'événements thrombotiques ou de coagulopathie de consommation (coagulation intravasculaire disséminée, CIVD) doit faire l'objet d'une surveillance clinique à l'aide de tests biologiques appropriés afin de détecter les signes précurseurs d'une coagulopathie thrombotique ou d'une coagulopathie de consommation. Dans ces cas, le bénéfice du traitement par ALPROLIX doit être mis en balance avec les risques de ces complications.
Perfusion continue
La sécurité et l'efficacité de l'utilisation d'APROLIX en perfusion continue ne sont pas démontrées (voir «Posologie/Mode d'emploi»).
Événements cardiovasculaires
Chez les patients présentant déjà des facteurs de risque cardiovasculaire, un traitement de substitution par des produits de facteur IX peut augmenter le risque cardiovasculaire.
Complications liées aux cathéters
Si un cathéter veineux central (CVC) est nécessaire, il convient de tenir compte du risque de complications associées au CVC, y compris 'le risque d'infections locales, de bactériémies et de thromboses au point d'insertion du cathéter.
Enfants et adolescents
Les avertissements et mesures de précaution indiqués valent aussi bien pour les adultes que pour les enfants.
Remarque concernant les excipients
Ce médicament contient moins de 1 mmol (23 mg) de sodium par flacon, c.-à-d. qu'il est essentiellement « sans sodium ». Il contient 0,3 mmol (soit 6,4 mg) de sodium par flacon. Il convient d'en tenir compte pour les patients devant veiller à suivre une alimentation pauvre en sodium.
Interactions
Il n'a été fait état d'aucune interaction entre 'ALPROLIX et d'autres médicaments. Aucune étude d'interaction n'a été menée.
Grossesse/Allaitement
Grossesse
Aucune étude de reproduction n'a été menée sur des animaux avec ALPROLIX. Une étude a été effectuée sur le transfert placentaire chez les souris (voir «Données précliniques»). L'hémophilie B 'étant rare chez les femmes, il n'existe aucune expérience sur l'utilisation du facteur IX pendant la grossesse et l'allaitement. Par conséquent, il convient de n'utiliser le facteur IX pendant la grossesse et l'allaitement qu'en cas de nécessité absolue.
Effet sur l’aptitude à la conduite et l’utilisation de machines
ALPROLIX n'a aucune influence sur l'aptitude à la conduite ou l'utilisation de machines.
Effets indésirables
Résumé du profil de sécurité
Dans de rares cas, en relation avec les traitements de substitution par des facteurs de coagulation, des réactions d'hypersensibilité ou des réactions allergiques sont possibles et peuvent comprendre des angio-œdèmes, une sensation de brûlure et de piqûre au site de perfusion, des frissons, une sensation de chaleur, une urticaire généralisée, des papules, une hypotonie, une léthargie, des nausées, une agitation, une tachycardie, une sensation d'oppression dans la poitrine, des fourmillements, des vomissements, des sifflements. Dans certains cas, ces réactions ont évolué en une grave anaphylaxie (avec choc) et ont été accompagnées de l'apparition d'inhibiteurs de facteur IX (voir également le paragraphe «Mises en garde et précautions»). Chez les patients atteints d'hémophilie B, il peut se former des anticorps neutralisants (inhibiteurs) contre le facteur IX. Ces inhibiteurs se manifestent sous la forme d'une réponse clinique insuffisante au médicament. Dans ce cas, il convient de contacter un centre spécialisé dans l'hémophilie. Après des essais d'induire une tolérance immunitaire chez les patients atteints d'hémophilie B avec des inhibiteurs du facteur IX et des réactions allergiques, des cas de syndrome néphrotique ont été signalés.
Après l'administration de préparations de facteur IX, il existe un risque d'épisodes thromboemboliques. Ce risque est plus élevé avec les produits ayant un faible degré de pureté. Lors de l'utilisation de facteur IX ultra-pur, les complications thromboemboliques sont rares.
Liste des effets indésirables
Patients atteints (PTPs): La sécurité d'ALPROLIX a été observé chez un total de 153 patients atteints d'hémophilie B sévère dans le cadre de deux études cliniques de phase III et d'une étude d'extension. Les événements indésirables ont été surveillés sur 561 sujets-années au total. Le nombre total de jours d'exposition a été de 26 106, avec une médiane de 165 (intervalle: 1 à 528) jours d'exposition par sujet.
Patients non traités antérieurement (PUP): un total de 33 patients atteints d'hémophilie B sévère ont été suivis dans le cadre d'un essai clinique. Les événements indésirables ont été surveillés sur un total de 57,51 patients-années. Le nombre total de jours d'exposition était de 2233, avec une valeur médiane de 76 (intervalle de 1 à 137) jours d'exposition par patient.
La sécurité Les effets indésirables les plus fréquemment signalés lors de la prise d'ALPROLIX étaient, avec une incidence de 1,3 %, les céphalées, la paresthésie orale et l'uropathie obstructive (PTPs), érythème au site d'injection, bien que Inhibition du facteur IX et hypersensibilité chez les PUPs.
Les effets indésirables sont indiqués conformément à la classification des systèmes d'organes MedDRA (SOC et termes préférés).
Les fréquences ont été estimées selon la convention suivante: très fréquent (≥1/10); fréquent (<1/100, ≥1/10); occasionnel (<1/1000, ≥1/100); rare (<1/10'000, ≥1/1000); très rare (<1/10'000), inconnu (la fréquence ne peut être estimée sur la base des données disponibles). Les effets indésirables suivants ont été signalés dans le cadre d'études cliniques ou après l'introduction au marché sur ALPROLIX.
Tableau 2: effets indésirables de ALPROLIX
Classification des systèmes d'organes MedDRA | Effets indésirables | Fréquences |
| Affections hématologiques et du système lymphatique | Inhibiteurs du factor IX | Fréquent 1 |
| Affections du système immunitaire | Hypersensibilité Anaphylaxie | Fréquent 1 Cas singuliers |
| Troubles du métabolisme et de la nutrition | Perte d'appétit | Occasionnel |
| Affections du système nerveux | Céphalées Vertiges Dysgueusie | Fréquent Occasionnel Occasionnel |
| Affections cardiaques | Palpitations | Occasionnel |
| Affections vasculaires | Hypotonie | Occasionnel |
| Affections gastro-intestinales | Paresthésie orale Mauvaise haleine | Fréquent Occasionnel |
| Affections du rein et des voies urinaires | Uropathie obstructive Hématurie Colique néphrétique Syndrome néphrotique (suite tentative tolérance immunitaire) | Fréquent Occasionnel Occasionnel Cas singuliers |
| Troubles généraux et anomalies au site d'administration | Érythème au site d'injection Fatigue Douleurs au site de perfusion | Fréquent Occasionnel Occasionnel |
1 La fréquence se réfère à l'occurrence dans l'étude avec les PUP. Les deux événements que sont l'inhibition du facteur IX et l'hypersensibilité sont survenus dans l'étude IV pour le même PUP. Voir la description d'effets indésirables sélectionnés
Description d'effets indésirables spécifiques
Dans l'ensemble du programme d'études cliniques, un patient (précédemment non traité) de l'étude IV a présenté un faible titre d'inhibiteurs du facteur IX associé à une hypersensibilité (voir «Efficacité clinique»). Après la commercialisation, le développement d'inhibiteurs du facteur IX et l'hypersensibilité (y compris l'anaphylaxie) ont été observés.
Enfants et adolescents
Chez l'enfant, les effets indésirables ont la même fréquence, la même nature et le même degré de sévérité que chez l'adulte.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
Aucun cas de surdosage n'a été rapporté à ce jour.
Propriétés/Effets
Code ATC
B02BD04
Mécanisme d'action
L'eftrenonacog alfa (facteur de coagulation IX humain recombinant, protéine de fusion Fc [rFIXFc]) possède 867 acides aminés. Il est fabriqué à l'aide de la 'technologie de l'ADN recombinant dans une lignée cellulaire embryonnaire rénale humaine (HEK) et sans adjonction de protéines exogènes, humaines ou animales, lors du processus de culture cellulaire, de purification ou de formulation finale.
Le facteur IX est une glycoprotéine à chaîne simple, avec une masse moléculaire d'env. 55000 Daltons. Le facteur de coagulation dépend de la vitamine K. Le facteur IX est activé par le facteur XIa dans le système de coagulation endogène et par le facteur VII / complexe facteur tissulaire dans le système de coagulation exogène. Le facteur IX activé, associé au facteur VIII activé, active le facteur X. Le facteur X activé transforme la prothrombine en thrombine. La thrombine transforme alors le fibrinogène en fibrine, et il se forme un caillot de sang.
L'hémophilie B est un trouble héréditaire, lié au sexe, de la coagulation sanguine, engendré par le faible taux de facteur IX fonctionnel. Elle entraîne des saignements dans les articulations, les muscles ou les organes internes, que ce soit de manière spontanée ou suite à un traumatisme chirurgical ou à un accident. La thérapie de substitution permet d'accroître le taux de facteur IX dans le plasma, ce qui permet de corriger provisoirement le déficit de facteur IX et la tendance hémorragique.
Pharmacodynamique
ALPROLIX (eftrenonacog alfa) est une protéine de fusion à action longue et entièrement recombinante, composée de facteur de coagulation IX humain, lié de manière covalente au domaine Fc de l'immunoglobine G1 humaine, et produit par la 'technologie de l'ADN recombinant.
La région Fc de l'immunoglobine G1 humaine se lie au récepteur Fc néonatal. Ce récepteur est exprimé pendant toute la vie du patient et fait partie d'une voie métabolique naturelle qui protège l'immunoglobine de la destruction lysosomale en réintégrant cette protéine dans la circulation sanguine, ce qui entraîne une longue demi-vie dans le plasma.
Efficacité clinique
La sécurité, l'efficacité et la pharmacocinétique d'ALPROLIX ont fait l'objet de deux études internationales ouvertes sur les patients atteints (PTPs) - une étude de phase 3 menée chez des adultes et des adolescents, appelée ici étude I, et une étude pédiatrique de phase 3, appelée étude II - (voir «Enfants et Adolescents»). La sécurité et l'efficacité d'ALPROLIX ont également été étudiées chez des patients (PUP) atteints d'hémophilie B sévère non traités auparavant (étude IV); voir «Enfants et Adolescents».
Dans le cadre de l'étude I, une comparaison de l'efficacité de deux régimes de traitement prophylactique (intervalle hebdomadaire fixe à une posologie de 50 UI/kg, et individualisé à une posologie de 100 UI/kg initialement tous les 10 jours) avec celle d'un traitement personnalisé a été effectuée. Cette étude a été conduite auprès d'un total de 123 patients de sexe masculin prétraités (12 à 71 ans) et atteints d'hémophilie B grave (activité FIX endogène ≤2%). Tous les patients ont reçu ALPROLIX et ont été suivis jusqu'à 77 semaines après le traitement.
Sur les 123 sujets qui ont achevé l'étude I, 93 ont été recrutés dans l'étude III (étude d'extension), avec une durée de suivi total médiane de 6,5 ans.
Il est à noter que le taux de saignement annualisé (Annualised Bleeding Rates, ABR) n'est pas comparable entre les différents concentrés de facteur, ni entre les différentes études cliniques.
Prophylaxie hebdomadaire fixe et individualisée:
La dose hebdomadaire médiane pour les sujets inclus dans le groupe hebdomadaire fixe était de 45,17 UI/kg (écart interquartile [EIQ]: 38,1-53,7) dans l'étude I. Parmi les sujets évaluables en termes d'efficacité, les taux de saignement annualisés ABR médians correspondants ont été de 2,95 (EIQ: 1,01-4,35) et sont restés du même ordre pendant toute la durée de l'étude III (1,85 [EIQ: 0,76-4,0]). Le taux médian de saignements articulaires spontanés a été de 0,38 (EIQ: 0,00-1,43) chez les sujets de l'étude III.
Chez les sujets dans le groupe individualisé, l'intervalle d'administration médian était de 12,53 jours (EIQ: 10,4-13,4) dans l'étude I l'ABR médian correspondant était de 1,38 (EIQ: 0,00-3,43) et est resté du même ordre pendant toute la durée de l'étude III (1,85 [EIQ: 0,76-4,0]).
Les intervalles d'administration et la consommation de facteur dans l'étude III (étude d'extension) sont restés comparables à ceux de l'étude I pour les deux protocoles de traitement prophylactique.
Aucun épisode hémorragique ne s'est produit chez 42 % des sujets sous prophylaxie individualisée et chez 23,0 % des sujets sous prophylaxie hebdomadaire. La proportion de sujets présentant ≥1 articulation cible à l'inclusion était plus faible dans le groupe de prophylaxie individualisée que dans le groupe de prophylaxie hebdomadaire (27,6 % et 57,1 %, respectivement).
Traitement des saignements:
Sur les 636 épisodes hémorragiques qui avaient été observés lors de l'étude I, 90,4 % ont été résolus avec 1 injection et 97,3 % avec 2 injections ou moins. La dose médiane par injection, en traitement d'un épisode hémorragique, était de 46,07 U.I./kg (EIQ: 32,86-57,03) UI/kg. La dose globale médiane utilisée pour traiter un épisode hémorragique a été de 51,47 UI/kg (EIQ: 35,21-61,73) dans le groupe de prophylaxie hebdomadaire, de 49,62 (EIQ: 35,71-94,82) dans le groupe de prophylaxie individualisée et de 46,58 (EIQ: 33,33-59,41) dans le groupe de traitement à la demande.
Prise en charge péri-opératoire (prophylaxie chirurgicale):
Au total, 35 interventions chirurgicales majeures ont été réalisées et évaluées chez 22 sujets (21 adultes et adolescents et 1 patient pédiatrique âgé de < 12 ans) dans l'étude I et dans l'étude III. Sur les 35 interventions chirurgicales majeures, 28 (80,0 %) ont nécessité une dose préopératoire unique pour maintenir l'hémostase pendant l'intervention chirurgicale. La médiane des doses moyennes par injection utilisée pour maintenir l'hémostase pendant l'intervention était de 94,7 UI/kg (intervalle: 49-152 UI/kg). La dose totale reçue le jour de l'intervention a été comprise entre 49 et 341 UI/kg, et la dose totale reçue pendant les 14 jours de la période péri-opératoire a été comprise entre 60 et 1 947 UI/kg.
La réponse hémostatique a été jugée excellente ou bonne dans 100 % des interventions chirurgicales majeures.
Enfants et adolescents
Lors de l'étude II, un total de 30 patients pédiatriques de sexe masculin prétraités et atteints d'hémophilie B grave (activité FIX endogène ≤2%) a été inclus. Les patients étaient âgés de moins de 12 ans (15 avaient moins de 6 ans et 15 avaient de 6 à 12 ans). Tous les patients ont reçu ALPROLIX et ont été suivis jusqu'à 52 semaines après le traitement.
Les 30 patients ont tous été traités par ALPROLIX selon un protocole de traitement prophylactique, en débutant à la dose de 50 - 60 UI/kg tous les 7 jours, avec possibilité d'ajustement de la dose jusqu'à un maximum de 100 UI/kg et de l'intervalle d'administration entre un minimum d'une fois par semaine et un maximum de deux fois par semaine. Parmi les 30 patients qui ont achevé l'étude II, 27 ont été recrutés dans l'étude III (étude d'extension). La durée médiane dans les études II + III était de 2,88 ans, avec un nombre moyen de 166 jours d'exposition.
L'étude IV a inclus 33 patients (PUP) de l'enfance et de l'adolescence atteints d'hémophilie B sévère (≤2 % d'activité FIX endogène) qui n'avaient jamais été traités auparavant. L'âge médian à l'inclusion dans l'étude était de 0,6 an (intervalle: 0,08 à 2 ans); 78,8 % des participants étaient âgés de moins d'un an. La durée médiane du traitement par ALPROLIX a été de 83,01 semaines au total (fourchette: 6,7 à 226,7 semaines), avec un nombre médian de jours d'exposition de 76 au total (fourchette: 1 à 137 jours).
Prophylaxie individualisée:
La médiane des doses hebdomadaires moyennes d'ALPROLIX était de 59,40 UI/kg (écart interquartile dans l'étude II: 52,95 à 64,78 UI/kg) chez les sujets âgés de < 6 ans et de 57,78 UI/kg (écart interquartile: 51,67 à 65,01 UI/kg) chez les sujets âgés de 6 à < 12 ans. Globalement, l'intervalle d'administration médian était de 6,99 jours (écart interquartile: 6,94 à 7,03) et ne s'est pas révélé différent selon la cohorte d'âge. À l'exception d'un patient chez lequel la dernière dose prescrite était de 100 UI/kg tous les 5 jours, les dernières doses prescrites chez les 29 autres patients étaient de 70 UI/kg maximum tous les 7 jours. Aucun épisode hémorragique ne s'est produit chez 33 % des sujets pédiatriques. Les intervalles d'administration et la consommation de facteur dans l'étude III sont restés comparables à ceux de l'étude II.
Les taux de saignement annualisés médians chez les sujets âgés de < 12 ans évaluables en termes d'efficacité étaient de 1,97 (écart interquartile: 0,00 - 3,13) dans l'étude II et sont restés du même ordre tout au long de l'étude III (étude d'extension).
Chez les PUP (étude IV), la dose hebdomadaire moyenne médiane d'ALPROLIX était de 57,96 UI/kg (intervalle interquartile de 52,45 à 65,06 UI/kg) et l'intervalle de dosage médian était de 7 jours (intervalle interquartile de 6,95 à 7,12 jours). Les intervalles de dosage et la consommation de facteurs étaient similaires dans l'étude IV et dans les études II et III. Parmi les PUP recevant une prophylaxie, 8 (28,6 %) n'ont pas connu d'épisodes hémorragiques. Le TBA médian de tous les participants sous prophylaxie était de 1,24 (écart interquartile de 0,0 à 2,49).
Traitement des épisodes hémorragiques:
Sur les 60 événements hémorragiques observés au cours de l'étude II, 75 % ont pu être contrôlés par 1 injection et, globalement, 91,7 % des événements hémorragiques ont pu être contrôlés par 2 injections ou moins. La médiane des doses moyennes par injection utilisée pour traiter un épisode hémorragique a été de 63,51 (écart interquartile: 48,92 à 99,44) UI/kg. La dose globale médiane utilisée pour traiter un épisode hémorragique a été de 68,22 UI/kg (écart interquartile: 50,89 à 126,19).
Sur les 58 épisodes hémorragiques chez des PUP sous prophylaxie dans l'étude IV, 87,9 % ont pu être contrôlés avec 1 injection et 96,6 % au total avec 2 injections ou moins. La dose médiane moyenne par injection pour traiter un épisode hémorragique était de 71,92 UI/kg (écart interquartile de 52,45 à 100,81 UI/kg). La dose totale médiane pour le traitement d'un épisode hémorragique était de 78,74 U.I./kg (intervalle interquartile de 53,57 à 104,90 U.I./kg).
Pharmacocinétique
Absorption
Toutes les études sur la pharmacocinétique d'ALPROLIX ont été menées auprès de patients atteints d'hémophilie B grave et ayant déjà reçu un traitement. Les données présentées dans ce paragraphe ont été recueillies à l'aide d'un test de coagulation en 1 étape, avec un réactif aPTT à base de silicium étalonné avec un facteur IX plasmatique témoin. Les propriétés pharmacocinétiques ont été évaluées auprès de 22 patients (≥19 ans) traités par ALPROLIX (rFIXFc). Le prélèvement des échantillons pharmacocinétiques a été effectué avant l'administration de 50 U.I./kg de rFIX (Benefix) ainsi qu'à 8 moments, sur une période allant jusqu'à 96 heures (4 jours) après l'administration. Après une phase de sevrage d'au moins 120 heures (5 jours), les patients ont reçu une dose unique de 50 U.I./kg d'ALPROLIX. Le prélèvement des échantillons pharmacocinétiques a été effectué 'avant l'administration, puis à 11 moments, sur une période allant jusqu'à 240 heures (10 jours) après l'administration. Les paramètres pharmacocinétiques après l'administration d'une dose de 50 U.I./kg d'ALPROLIX sont indiqués dans le tableau 3.
Tableau 3: paramètres pharmacocinétiques d'ALPROLIX (50 U.I./kg)
Paramètres pharmacocinétiques1 | ALPROLIX |
N=22 | |
Récupération incrémentielle (U.I./dl par U.I./kg) | 0,92 |
ASC/Dose (U.I.*h/dl par U.I./kg) | 31.32 |
Cmax (U.I./dl) | 40,81 |
CL (ml/h/kg) | 3,19 |
t1/2α (h) | 5,03 |
t1/2β (h) | 82,12 |
MRT (h) | 98,60 |
Vss (ml/kg) | 314,80 |
Temps jusqu'à 1% (en jours) | 11,22 (10,20-12,35) |
1 Les paramètres pharmacocinétiques sont indiqués sous forme de moyennes géométriques (IC à 95%)
Abréviations: IC = intervalle de confiance; Cmax = activité maximale; ASC = aire sous la courbe de l'activité FIX en fonction du temps; t1/2α = demi-vie de répartition;
t1/2β = demi-vie d'élimination; CL = clairance; Vss = volume de distribution à l'équilibre; MRT = durée de séjour moyenne.
La demi-vie d'élimination d'ALPROLIX (82 heures) est influencée par la région Fc déterminée de manière avérée dans les modèles animaux par le métabolisme du récepteur Fc néonatal.
Un modèle pharmacocinétique de population a été développé. Il repose sur les données de l'activité FIX de 161 patients de tous les âges (2 à 76 ans) pesant entre 12,5 et 186,7 kg et ayant participé à trois études cliniques (12 patients dans une étude de phase 1/2a, ainsi que 123 patients dans l'étude I et 26 patients dans l'étude II). La valeur attendue en termes de clairance d'ALPROLIX pour un adulte typique de 70 kg est de 2,30 dl/h et le volume de distribution à l'équilibre est de 194,8 dl pour ALPROLIX. Le modèle a été appliqué pour la prévision du profil activité-temps après une administration unique d'ALPROLIX chez des patients atteints d'hémophilie B grave (voir tableau 4).
Tableau 4: activité FIX prédite [U.I./dl] après une administration unique d'ALPROLIX1 chez des patients âgés de ≥12 ans
Dose (I.E./kg) | Fin de l'injection | 12 heures | 24 heures (jour 1) | 36 heures | 48 heures (jour 2) | 72 heures (jour 3) | Jour 5 | Jour 7 | Jour 10 | Jour 14 | |
Médiane (5è, 95è) | |||||||||||
50 | 52,0 | 21,7 | 15,2 | 11,3 | 8,4 | 5,5 | 3,02 | 1,93 | 1,07 | 0,495 | |
[31,8; 82] | [15,0; 31,0] | [10,2; 21,5] | [7,50; 16,5] | [5,5; 12,5] | [3,59; 8,25] | [1,88; 4,65] | [0.99; 3.12] | [0,345; 1,95] | [0,0829; 1,17] | ||
100 | 104,0 | 43,4 | 30,4 | 22,6 | 16,8 | 11 | 6,03 | 3,85 | 2,13 | 0,991 | |
[63,6; 164] | [30,0; 62,0] | [20,4; 42,9] | [15,0; 32,9] | [11; 24,9] | [7,18; 16,5] | [3,77; 9,29 | [1,98; 6,24] | [0,691; 3,89] | [0,166; 2,34] | ||
1 Voir «Posologie/Mode d'emploi»
Distribution
Voir «Absorption».
Métabolisme
Voir «Absorption».
Élimination
Voir «Absorption».
Cinétique pour certains groupes de patients
Enfants et adolescents
Les paramètres pharmacocinétiques d'ALPROLIX ont été déterminés chez les adolescents au cours de l'étude I et chez les enfants au cours de l'étude II (le prélèvement des échantillons pharmacocinétiques a été effectué avant l'administration et à plusieurs moments, sur une période allant jusqu'à 336 heures (14 jours) après l'administration dans l'étude I / avant l'administration et à 7 moments sur une période allant jusqu'à 168 heures (7 jours) après l'administration dans l'étude II.
Le tableau 5 présente les paramètres pharmacocinétiques qui ont été calculés sur la base des données pédiatriques de 35 patients âgés de moins de 18 ans.
Tableau 5: comparaison des paramètres pharmacocinétiques d'ALPROLIX (rFIXFc) par classe d'âge
Paramètres pharmacocinétiques1 | Étude II | Étude I | |||
<6 ans (intervalle 2-4) | 6 à <12 ans (intervalle 6-10) | 12 à <18 ans (intervalle 12-17) | |||
N = 11 | N = 13 | N = 11 | |||
IR (U.I./dl par U.I./kg) | 0,5898 | 0,7170 | 0,8470 | ||
ASC/Dose (U.I.*h/dl par U.I./kg) | 22,71 | 28,53 | 29,50 | ||
t½ (h) | 66,49 | 70,34 | 82,22 | ||
MRT (h) | 83,65 | 82,46 | 93,46 | ||
CL (ml/h/kg) | 4,365 | 3,505 | 3,390 | ||
Vss (ml/kg) | 365,1 | 289,0 | 316,8 | ||
1Les paramètres pharmacocinétiques de l'analyse non compartimentale sont indiqués sous forme de moyennes géométriques (IC à 95%)
Abréviations: IC = intervalle de confiance; IR = récupération incrémentielle; ASC = aire sous la courbe de l'activité FIX en fonction du temps; t1/2 = demi-vie terminale; MRT = durée de séjour moyenne; CL = clairance; Vss = volume de distribution à l'équilibre
Données précliniques
Sur la base des études de toxicité aiguë et de toxicité en cas d'administration répétée, qui incluaient les évaluations de la toxicité locale et de la pharmacologie de sécurité, les données précliniques ne permettent pas d'identifier de risque particulier pour l'être humain. Des études axées sur la génotoxicité, la cancérogénicité, la toxicité reproductive ou le développement embryonnaire et fœtal n'ont pas été menées. Une étude sur le transfert plasmatique a démontré que chez la souris, Eftrenonacog alfa (rFIXFc) passent la barrière placentaire.
Fertilité
Aucune étude de fertilité n'a été menée sur des animaux avec ALPROLIX.
Remarques particulières
Incompatibilités
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé à d'autres médicaments.
N'utiliser que le kit d'injection fourni, car certains équipements d'injection risquent, en raison d'une adsorption du facteur de coagulation IX sur les surfaces intérieures, de causer l'échec du traitement.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage. Pendant la durée de conservation, le médicament peut être entreposé, pendant une période unique de 6 mois maximum, à température ambiante (jusqu'à 30°C). Noter sur l'emballage la date à laquelle le médicament est sorti du réfrigérateur. Après la conservation du médicament à température ambiante, ne pas le replacer au réfrigérateur.
Après la reconstitution
La stabilité chimique et physique a été montrée pour 6 heures à température ambiante (jusqu'à 30°C). Si le médicament n'est pas utilisé dans les 6 heures, l'éliminer. Pour des raisons microbiologiques, le médicament doit être utilisé immédiatement après sa reconstitution. Si cela s'avère impossible, les délais d'utilisation et les conditions de conservation relèvent de la responsabilité de l'utilisateur. Protéger le médicament des rayons directs du soleil.
Remarques particulières concernant le stockage
Conserver hors de portée des enfants.
Conserver le récipient dans son carton pour le protéger de la lumière. Conserver au réfrigérateur (2-8°C). Ne pas congeler. Ce médicament est destiné à une utilisation unique. Les restes de solution doivent être éliminés correctement.
Conditions de conservation après reconstitution du médicament: voir «Stabilité».
Remarques concernant la manipulation
Le contenu du flacon contenant la poudre lyophilisée pour solution injectable doit être reconstituée à l'aide du solvant fourni (solution de chlorure de sodium) dans la seringue préremplie, en utilisant l'adaptateur stérile pour flacons. Incliner légèrement le flacon dans un sens et dans l'autre jusqu'à ce que la solution soit entièrement dissoute.
La solution reconstituée doit être claire à légèrement irisée et incolore. Le médicament reconstitué doit être inspecté visuellement avant l'administration pour vérifier l'absence de particules et de décoloration. La solution ne doit pas être utilisée si elle est trouble ou présente des dépôts.
Ce produit est à usage unique. ALPROLIX ne doit pas être mélangé avec d'autres solutions injectables ou pour perfusion.
Le médicament non utilisé ou les déchets doivent être éliminés de manière appropriée.
Un emballage d'ALPROLIX contient:
| A) 1 flacon de poudre |
Préparation:
Avant l'ouverture du flacon, se laver les mains
1. Vérifiez le nom et le dosage indiqués sur l'emballage afin de vous assurer qu'il contient le bon produit. Vérifiez la date de péremption sur le carton extérieur d'ALPROLIX. Ne pas utiliser si la date de péremption du médicament est dépassée. | |
2. Si ALPROLIX a été conservé dans un réfrigérateur, attendez que le flacon d'ALPROLIX (A) et la seringue contenant le solvant (B) soient à température ambiante avant de les utiliser. N'utilisez pas de source de chaleur externe. | |
3. Placez le flacon sur une surface propre et plane. Retirez le bouchon en plastique du flacon.. |
|
4. Nettoyez le haut du flacon à l'aide d'un des tampons d'alcool (F) fournis dans l'emballage et laissez-le sécher à l'air. Après l'avoir essuyé, évitez tout contact avec la partie supérieure du flacon ou tout autre objet. |
|
5. Retirez le papier de protection de l'adaptateur en plastique transparent (D). Ne retirez pas l'adaptateur de son capuchon protecteur. Évitez de toucher l'intérieur de l'emballage de l'adaptateur de flacon. | |
6.Placez le flacon sur une surface plane. Tenez l'adaptateur du flacon par son capuchon protecteur et placez-le tout droit sur le haut du flacon. Poussez fermement l'adaptateur vers le bas jusqu'à ce qu'il s'enclenche en haut du flacon et que le mandrin de l'adaptateur traverse le bouchon du flacon. |
|
7. Raccordez la tige du piston (C) à la seringue de solvant en insérant l'extrémité de la tige du piston dans l'ouverture du piston de la seringue. Tournez vigoureusement la tige de piston dans le sens des aiguilles d'une montre jusqu'à ce qu'elle soit bien fixée dans le piston de la seringue. |
|
8. Cassez le capuchon en plastique inviolable blanc de la seringue de solvant en le pliant vers le bas au niveau de la perforation jusqu'à ce qu'il se brise. Posez le capuchon sur une surface plane, face vers le bas. Ne touchez pas l'intérieur du capuchon ou l'embout de la seringue. |
|
9. Retirez le capuchon de protection de l'adaptateur et jetez-le. |
|
10. Connectez la seringue de solvant à l'adaptateur de flacon en insérant l'extrémité de la seringue dans l'ouverture de l'adaptateur. Appuyez et tournez fermement la seringue dans le sens des aiguilles d'une montre jusqu'à ce qu'elle soit bien fixée. |
|
11. Poussez lentement la tige du piston vers le bas pour injecter la totalité du solvant dans le flacon d'ALPROLIX. |
|
12. Laisser la seringue sur l'adaptateur et la tige du piston abaissée et agiter doucement le flacon jusqu'à ce que la poudre soit dissoute. Ne pas agiter. |
|
13. La solution finale doit être contrôlée visuellement avant l'administration. La solution doit être claire à légèrement irisée et incolore. Elle ne doit pas être utilisée si elle est trouble ou si elle contient des particules visibles. | |
14. Veillez à ce que la tige du piston soit toujours complètement enfoncée dans la seringue, puis retournez le flacon. Retirez lentement la tige du piston pour aspirer toute la solution dans la seringue à travers l'adaptateur du flacon. Remarque: si vous utilisez plus d'un flacon d'ALPROLIX par injection, préparez chaque flacon séparément en suivant les instructions ci-dessus (étapes 1 à 13). Retirez la seringue de solvant et laissez l'adaptateur de flacon connecté. Une seule grande seringue Luer Lock peut être utilisée pour aspirer les solutions préparées de chacun des flacons individuels. |
|
15. Retirez la seringue de l'adaptateur de flacon en tirant doucement sur le flacon et en le tournant dans le sens inverse des aiguilles d'une montre. |
|
16. Jetez le flacon et l'adaptateur. Remarque: si vous n'utilisez pas la solution immédiatement, le capuchon de la seringue doit être replacé avec précaution sur l'embout de la seringue. Ne touchez pas l'embout de la seringue ou l'intérieur du capuchon. Une fois préparé, ALPROLIX peut être conservé à température ambiante pendant 6 heures au maximum avant d'être administré. Après cette période, ALPROLIX préparé doit être éliminé. Protéger de la lumière directe du soleil. | |











Administration:
1.Ouvrez l'emballage contenant le kit de perfusion et retirez le capuchon à l'extrémité de la tubulure. Raccordez la seringue contenant la solution ALPROLIX préparée à l'extrémité de la tubulure de perfusion en la tournant dans le sens des aiguilles d'une montre. |
|
2.Si nécessaire, utilisez un garrot et préparez le site d'injection en essuyant soigneusement la peau avec le deuxième tampon d'alcool de l'emballage.
| |
3. Retirez tout l'air de la tubulure de perfusion en appuyant lentement sur la tige du piston jusqu'à ce que le liquide atteigne l'aiguille du dispositif de perfusion. Ne faites pas passer la solution à travers l'aiguille. Retirez la gaine de protection en plastique transparent de l'aiguille. | |
4. Insérez l'aiguille du dispositif de perfusion dans une veine, comme indiqué par votre médecin ou votre professionnel de santé, et retirez le garrot. Si vous le souhaitez, vous pouvez utiliser l'un des pansements (G) de l'emballage pour fixer les ailettes en plastique de l'aiguille au site d'injection. Le médicament préparé doit être injecté par voie intraveineuse pendant plusieurs minutes. Il est possible que votre médecin modifie la vitesse d'injection recommandée pour vous afin de vous rendre la tâche plus confortable. | |
5. Une fois l'injection terminée et l'aiguille retirée, rabattez le protège-aiguille sur l'aiguille et laissez-le s'enclencher. |
|
6.Jeter l'aiguille usagée, la solution non utilisée, la seringue et le flacon vide de manière sûre dans un conteneur approprié pour les déchets médicaux, car ces matériaux peuvent blesser d'autres personnes s'ils ne sont pas éliminés de manière appropriée. Le matériel ne doit pas être réutilisé. | |
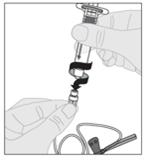


Numéro d’autorisation
66039 (Swissmedic)
Titulaire de l’autorisation
Swedish Orphan Biovitrum AG, Basel
Mise à jour de l’information
décembre 2021
Отзывов (0)

Бесплатная консультация опытного специалиста
Опишите симптомы или нужный препарат – мы поможем подобрать его дозировку или аналог, оформим заказ с доставкой на дом или просто проконсультируем.
Нас 14 специалистов и 0 ботов. Мы всегда будем с вами на связи и сможем связаться в любое время.