




TALTZ Inj Lös 80 мг/мл предварительно заполненный шприц
Taltz Inj Lös 80 mg/ml Fertigspritze
-
239104.52 RUB

- Наличие: Нет в наличии
- Производитель: ELI LILLY (SUISSE) SA
- Модель: 6939959
- ATC-код L04AC13
- EAN 7680659060018
Состав:
Наведите телефон на qr-код

Описание
 Deutsch
Deutsch French
French Italian
ItalianWas ist Taltz und wann wird es angewendet?
Taltz enthält den Wirkstoff Ixekizumab. Ixekizumab ist ein monoklonaler Antikörper. Monoklonale Antikörper sind Eiweisse, die bestimmte Eiweisse im Körper erkennen und an sie binden.
Taltz gehört zu einer Gruppe von Arzneimitteln, die als Interleukin-Inhibitoren (IL-Inhibitoren) bezeichnet werden. Dieses Arzneimittel wirkt durch Neutralisieren der Aktivität eines Interleukins, das IL-17A genannt wird und bei Erkrankungen wie Plaque-Psoriasis und Psoriasis-Arthritis in erhöhten Konzentrationen vorliegt.
Plaque-Psoriasis
Taltz wird auf Verschreibung des Arztes/der Ärztin zur Behandlung einer als «Plaque-Psoriasis» bezeichneten Hauterkrankung angewendet, die von einer die Haut betreffenden Entzündung hervorgerufen ist. Taltz mindert die Entzündung und andere Symptome der Erkrankung, wie Schuppung, Jucken und Schmerzen.
Taltz wird bei Erwachsenen und bei Kindern und Jugendlichen (ab 6 Jahren, mit einem Körpergewicht von mindestens 25 kg) mit mittelschwerer bis schwerer Plaque-Psoriasis angewendet, die auf andere Therapien nicht angesprochen haben, bei denen diese Therapien kontraindiziert sind oder die diese Therapien nicht tolerieren.
Psoriasis-Arthritis
Taltz wird zur Behandlung einer als «Psoriasis-Arthritis» bezeichneten Erkrankung angewendet. Es handelt sich um eine entzündliche Erkrankung der Gelenke, die oft von einer Hauterkrankung, Plaque-Psoriasis, begleitet wird.
Taltz reduziert die Anzeichen und Symptome der Krankheit und verbessert die körperliche Funktion (Fähigkeit, normale tägliche Aktivitäten zu tun).
Taltz wird auf Verschreibung des Arztes/der Ärztin, alleine oder zusammen mit einem anderen Arzneimittel (Antirheumatikum), bei erwachsenen Patienten mit Psoriasis-Arthritis angewendet, die auf herkömmliche Therapie nicht ausreichend angesprochen haben.
Ankylosierende Spondylitis Taltz wird zur Behandlung einer als «ankylosierende Spondylitis» bezeichneten Erkrankung angewendet. Es handelt sich um eine entzündliche Krankheit, die vor allem die Wirbelsäule betrifft und eine Entzündung der Wirbelgelenke verursacht.
Taltz wird auf Verschreibung des Arztes/der Ärztin, alleine oder zusammen mit anderen Arzneimitteln zur Behandlung dieser Krankheit, bei erwachsenen Patienten mit ankylosierender Spondylitis angewendet, die auf herkömmliche Therapie nicht ausreichend angesprochen oder diese nicht vertragen haben.
Wann darf Taltz nicht angewendet werden?
Wenden Sie Taltz nicht an, wenn Sie auf den Wirkstoff Ixekizumab oder einen der in Taltz enthaltenen Hilfsstoffe (siehe «Was ist in Taltz enthalten?») überempfindlich sind.
Taltz darf nicht verabreicht werden, wenn Sie an einer schweren Infektion leiden, wie z.B. Tuberkulose.
Wann ist bei der Anwendung von Taltz Vorsicht geboten?
Fragen Sie Ihren Arzt/Apotheker oder Ihre Ärztin/Apothekerin, bevor Sie Taltz anwenden:
- wenn Sie derzeit eine Infektion haben oder an einer chronischen Infektion oder an wiederholten Infektionen leiden.
- wenn Sie Tuberkulose haben.
- wenn Sie an einer entzündlichen Erkrankung des Darms (wie Morbus Crohn oder Colitis ulzerosa) leiden. Bitte informieren Sie sofort Ihren Arzt bzw. Ihre Ärztin wenn Sie Bauchschmerzen, Bauchkrämpfe, Durchfall, Gewichtsverlust oder Blut im Stuhl haben.
- wenn Sie kürzlich eine Impfung erhalten haben oder während der Behandlung mit Taltz eine Impfung planen.
- wenn Sie eine andere Plaque-Psoriasis- oder Psoriasis-Arthritis-Therapie erhalten (Arzneimittel oder ultraviolettes (UV) Licht).
Taltz kann möglicherweise schwerwiegende Nebenwirkungen haben, hierzu gehören Infektionen und allergische Reaktionen. Während der Anwendung von Taltz müssen Sie auf Symptome dieser Erkrankungen achten. Wenn Sie die folgenden Anzeichen feststellen, die auf eine mögliche schwerwiegende Infektion oder eine allergische Reaktion hinweisen, beenden Sie sofort die Anwendung von Taltz und informieren Sie Ihren Arzt bzw. Ihre Ärztin oder suchen Sie medizinische Hilfe auf.
Anzeichen einer potenziell schwerwiegenden Infektion: Fieber, grippeartige Symptome, nächtliches Schwitzen, Müdigkeitsgefühl oder Kurzatmigkeit; Husten, der nicht vorübergeht; warme, gerötete und schmerzhafte Haut oder schmerzhafter Hautausschlag mit Bläschen, Brennen beim Wasserlassen.
Anzeichen einer allergischen Reaktion: Schwierigkeiten beim Atmen oder Schlucken, niedriger Blutdruck, der Schwindel oder Benommenheit hervorrufen kann; Schwellung des Gesichts, der Lippen, des Mundes oder des Halses.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro 80 mg Dosis, d.h. es ist nahezu «natriumfrei».
Informieren Sie Ihren Arzt/Apotheker bzw. Ihre Ärztin/Apothekerin, wenn Sie an anderen Krankheiten leiden, Allergien haben oder andere Arzneimittel (auch selbst gekaufte!) einnehmen/anwenden.
Darf Taltz während einer Schwangerschaft oder in der Stillzeit angewendet werden?
Die Verwendung von Taltz in der Schwangerschaft sollte vermieden werden. Die Wirkungen des Arzneimittels bei Schwangeren sind nicht bekannt. Sprechen Sie mit Ihrem Arzt bzw. Ihrer Ärztin, wenn Sie schwanger sind, glauben, schwanger zu sein, oder schwanger werden möchten. Während der Behandlung mit Taltz und mindestens 10 Wochen nach der letzten Gabe von Taltz ist der Eintritt einer Schwangerschaft unter Anwendung von geeigneten Verhütungsmassnahmen zu vermeiden.
Es ist nicht bekannt, ob der Wirkstoff von Taltz in die Muttermilch übertritt. Wegen möglicher Schäden für den gestillten Säugling wird empfohlen, während der Behandlung mit Taltz und mindestens 10 Wochen nach der letzten Dosis nicht zu stillen. Sprechen Sie vor der Anwendung von Taltz mit Ihrem Arzt bzw. Ihrer Ärztin, wenn Sie stillen oder stillen möchten. Sie und Ihr Arzt bzw. Ihre Ärztin müssen entscheiden, ob Sie stillen oder Taltz anwenden werden. Beides zusammen dürfen Sie nicht.
Wie verwenden Sie Taltz?
Wenden Sie Taltz immer genau nach Anweisung Ihres Arztes oder Apothekers bzw. Ihrer Ärztin oder Apothekerin oder des Pflegepersonals an. Bitte fragen Sie bei Ihrem Arzt oder Apotheker bzw. Ihrer Ärztin oder Apothekerin oder bei Ihrem Pflegepersonal nach, wenn Sie sich nicht ganz sicher sind, wie das Arzneimittel angewendet wird.
Taltz wird als Injektion unter die Haut verabreicht (subkutan). Sie und Ihr Arzt bzw. Ihre Ärztin entscheiden, ob Sie sich Taltz selbst injizieren sollen.
Es ist wichtig, dass Sie nicht versuchen, sich selbst zu injizieren, bevor Sie durch Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin oder das Pflegepersonal ein Training erhalten haben. Eine Pflegeperson kann Ihnen ebenfalls nach entsprechendem Training Ihre Taltz-Injektion verabreichen.
Lesen Sie die Bedienungsanleitung für die Fertigspritze sorgfältig durch, bevor Sie Taltz verwenden.
Wie viel Taltz wird angewendet und für wie lange?
Jede Fertigspritze ist für den einmaligen Gebrauch bestimmt und gibt eine Taltz-Dosis von 80 mg ab. Die vollständige Menge der Fertigspritze soll injiziert werden.
Plaque Psoriasis bei Erwachsenen
Bei der ersten Behandlung werden 160 mg (zwei 80 mg Injektionen) als subkutane Injektion verabreicht. Diese werden Sie durch Ihren Arzt bzw. Ihre Ärztin oder durch das Pflegepersonal erhalten.
Nach der ersten Behandlung werden Sie in den Wochen 2, 4, 6, 8, 10 und 12, danach alle 4 Wochen eine 80 mg Dosis (eine Injektion) erhalten.
Wenn Ihr Körpergewicht unter 100 kg liegt, wird Ihr Arzt bzw. Ärztin entscheiden, ob Sie eine alternative Dosierung erhalten und ab Woche 2 Ihnen eine Dosis von 80 mg alle 4 Wochen verabreicht wird.
Plaque-Psoriasis bei Kindern und Jugendlichen (ab 6 Jahren)
Kinder und Jugendliche ab 6 Jahren mit einem Körpergewicht über 50 kg:
Zu Beginn der Behandlung wird eine einmalige Initialdosis von 160 mg (zwei Injektionen zu 80 mg) als subkutane Injektion verabreicht. Ab der 4. Woche nach der ersten Behandlung und danach alle 4 Wochen wird eine 80 mg Dosis (eine Injektion zu 80 mg) verabreicht.
Kinder und Jugendliche ab 6 Jahren mit einem Körpergewicht von 25 kg bis 50 kg:
Zu Beginn der Behandlung wird eine einmalige Initialdosis von 80 mg (eine Injektion zu 80 mg) als subkutane Injektion verabreicht. Ab der 4. Woche nach der ersten Behandlung und danach alle 4 Wochen wird eine 40 mg Dosis verabreicht.
Der Arzt bzw. die Ärztin oder das Pflegepersonal wird Ihrem Kind Taltz verabreichen.
Für die 80 mg Dosis soll die Gesamtmenge der Fertigspritze verabreicht werden.
40 mg Dosen stehen nicht als Fertigspritze zur Verfügung und müssen von einer qualifizierten medizinischen Fachperson vorbereitet werden (siehe «Was ist ferner zu beachten?»).
Psoriasis-Arthritis
Bei der ersten Behandlung werden 160 mg (zwei 80 mg Injektionen) als subkutane Injektion verabreicht. Diese werden Sie durch Ihren Arzt bzw. Ihre Ärztin oder durch das Pflegepersonal erhalten.
Nach der ersten Behandlung werden Sie alle 4 Wochen eine 80 mg Dosis (eine Injektion) erhalten.
Wenn Sie zusätzlich zu Psoriasis-Arthritis auch an mittelschwerer bis schwerer Plaque-Psoriasis leiden, wird Ihnen Ihr Arzt bzw. Ihre Ärztin die Dosierung für Plaque-Psoriasis verschreiben.
Ankylosierende Spondylitis
Die empfohlene Dosis beträgt 80 mg (eine Injektion) als subkutane Injektion alle 4 Wochen.
Während der Behandlung mit Taltz wird Ihr Arzt bzw. Ihre Ärztin Ihnen gegebenenfalls weiterhin andere konventionelle krankheitsmodifizierende Antirheumatika verschreiben, beispielsweise Sulfasalazin, Kortikosteroide, nichtsteroidale entzündungshemmende Medikamente und Analgetika.
Taltz ist für die Langzeittherapie bestimmt. Ihr Arzt bzw. Ihre Ärztin wird Ihren Krankheitsverlauf regelmässig kontrollieren, um zu überprüfen, ob die Behandlung die gewünschte Wirkung hat.
Wenn Sie eine grössere Menge von Taltz angewendet haben, als Sie sollten
Wenn Sie versehentlich mehr Taltz angewendet haben oder die Dosis früher gegeben wurde, als Ihnen von Ihrem Arzt bzw. Ihrer Ärztin verschrieben wurde, informieren Sie Ihren Arzt bzw. Ihre Ärztin.
Wenn Sie die Anwendung von Taltz vergessen haben
Wenn Sie vergessen haben, eine Taltz-Dosis zu injizieren, holen Sie dies nach, sobald Sie daran denken. Dann sprechen Sie mit Ihrem Arzt bzw. Ihrer Ärztin, wann Sie die nächste Dosis injizieren sollen.
Kinder und Jugendliche
Die Erfahrung bei Kindern und Jugendlichen mit Plaque-Psoriasis ab 6 Jahren ist über 1 Jahr Therapielänge limitiert. Taltz wird bei Kindern und Jugendlichen unter 6 Jahren bei der Behandlung von Plaque-Psoriasis nicht empfohlen, da die Anwendung in dieser Altersgruppe nicht geprüft wurde.
Taltz wird bei Kindern und Jugendlichen unter 18 Jahren bei der Behandlung von Psoriasis-Arthritis und ankylosierende Spondylitis nicht empfohlen, da die Anwendung in dieser Altersgruppe nicht geprüft wurde.
Halten Sie sich an die in der Packungsbeilage angegebene oder vom Arzt oder der Ärztin verschriebene Dosierung. Wenn Sie glauben, das Arzneimittel wirke zu schwach oder zu stark, so sprechen Sie mit Ihrem Arzt oder Apotheker bzw. mit Ihrer Ärztin oder Apothekerin.
Welche Nebenwirkungen kann Taltz haben?
Schwerwiegende Nebenwirkungen
Brechen Sie die Anwendung von Taltz ab und sprechen Sie sofort mit Ihrem Arzt/Ihrer Ärztin bzw. suchen Sie ärztliche Hilfe auf, wenn Sie eine der folgenden Nebenwirkungen bemerken. Ihr Arzt bzw. Ihre Ärztin wird entscheiden, ob und wann Sie die Therapie wiederbeginnen können:
Mögliche schwerwiegende Infektion: Zu den Anzeichen können gehören:
- Fieber, grippeartige Symptome, nächtliches Schwitzen;
- Müdigkeitsgefühl oder Kurzatmigkeit, Husten, der nicht vorübergeht;
- warme, gerötete und schmerzhafte Haut oder schmerzhafter Hautausschlag mit Bläschen.
Schwerwiegende allergische Reaktion: Zu den Anzeichen können gehören:
- Schwierigkeiten beim Atmen oder Schlucken;
- Schwellung von Gesicht, Lippen, Zunge oder Hals;
- schweres Jucken der Haut, mit rotem Ausschlag oder Erhebungen (Nesselsucht).
Folgende Nebenwirkungen können bei der Anwendung von Taltz auftreten. Die meisten verlaufen leicht oder mittelschwer.
Sehr häufig (betrifft mehr als einen von 10 Anwendern)
Infektionen der oberen Atemwege mit Symptomen wie Halsschmerzen und verstopfter Nase (Nasopharyngitis), Reaktionen an der Injektionsstelle (z.B. gerötete Haut, Schmerzen).
Häufig (betrifft 1 bis 10 von 100 Anwendern)
Übelkeit, Durchfall, Pilz-Infektionen der Haut, Schmerzen im hinteren Halsbereich.
Gelegentlich (betrifft 1 bis 10 von 1000 Anwendern)
Pilzinfektion im Mundraum, Grippe, laufende Nase, Nesselsucht, Augenausfluss mit Juckreiz Rötung und Schwellung (Konjunktivitis), Anzeichen einer geringen Anzahl weisser Blutkörperchen (Neutropenie) wie Fieber, Halsschmerzen oder Mundgeschwüre infolge von Infektionen, niedrige Zahl der Blutplättchen (Thrombozytopenie), entzündliche Darmerkrankung (Morbus Crohn, Colitis ulzerosa, siehe «Wann ist bei der Anwendung von Taltz Vorsicht geboten?»).
Selten (betrifft 1 bis 10 von 10'000 Anwendern)
Pilzinfektion in der Speiseröhre, anaphylaktische (allergische) Reaktion mit Anzeichen wie Schwellung von Gesicht, Lippen, Zunge oder Hals und Nesselsucht (siehe «Schwerwiegende allergische Reaktion» oben).
Berichtete Nebenwirkungen bei Kindern und Jugendlichen sind insgesamt vergleichbar mit denen bei Erwachsenen.
Grippe, Nesselsucht, Augenausfluss mit Juckreiz, Rötung und Schwellung (Konjunktivitis), Anzeichen einer geringen Anzahl weisser Blutkörperchen (Neutropenie) wie Fieber, Halsschmerzen oder Mundgeschwüre infolge von Infektionen, und Überempfindlichkeitsreaktionen (siehe «Schwerwiegende Nebenwirkungen», «Schwerwiegende allergische Reaktion» oben) wurden jedoch häufig gemeldet.
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Dies gilt insbesondere auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind.
Was ist ferner zu beachten?
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Lagerungshinweis
Im Kühlschrank (2-8°C) lagern. In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Nicht einfrieren. Taltz, das eingefroren war, darf nicht verwendet werden.
Temporäre Lagerung: wenn Kühlung nicht möglich ist, können Sie Ihre Fertigspritze bis zu insgesamt 5 Tagen bei Temperaturen unter 30°C aufbewahren.
Ausser Reichweite von Kindern aufbewahren.
Weitere Hinweise
Jede Fertigspritze ist zum einmaligen Gebrauch bestimmt.
Nicht schütteln.
Nicht verwenden, wenn Partikel auftreten oder wenn die Lösung wolkig und/oder merklich braun ist.
Die Bedienungsanleitung der Fertigspritze muss sorgfältig beachtet werden.
Weitere Auskünfte erteilt Ihnen Ihr Arzt oder Apotheker, bzw. Ihre Ärztin oder Apothekerin. Diese Personen verfügen über die ausführliche Fachinformation.
Taltz 40 mg Dosen für pädiatrische Patienten von 25 kg bis zu 50 kg dürfen nur von medizinischem Fachpersonal, gemäss Fachinformation, vorbereitet werden.
Was ist in Taltz enthalten?
Wirkstoffe
Jede Fertigspritze enthält 80 mg Ixekizumab als Wirkstoff in 1 ml Injektionslösung.
Hilfsstoffe
Natriumcitrat-Dihydrat, Zitronensäure, Natriumchlorid, Polysorbat 80, Wasser für Injektionszwecke.
Zulassungsnummer
65906 (Swissmedic).
Wo erhalten Sie Taltz? Welche Packungen sind erhältlich?
In Apotheken nur gegen ärztliche Verschreibung.
Packungen mit 1 und 2 Fertigspritzen.
Zulassungsinhaberin
Eli Lilly (Suisse) SA, 1214 Vernier/GE.
Diese Packungsbeilage wurde im Juli 2020 letztmals durch die Arzneimittelbehörde (Swissmedic) geprüft.
Bedienungsanleitung
Taltz® Ixekizumab 80mg Injektionslösung in einer Fertigspritze |
|
Bitte lesen Sie die Packungsbeilage (Patienteninformation) und diese Bedienungsanleitung VOLLSTÄNDIG durch, bevor Sie Ihre Taltz Fertigspritze benutzen. |
BEVOR SIE IHRE FERTIGSPRITZE BENUTZEN: WICHTIG ZU WISSEN |
|
|

Beschreibung der Fertigspritze |
|

1 | BEVOR SIE ANFANGEN | |
1a | Nehmen Sie die Fertigspritze aus dem Kühlschrank. Lassen Sie die Nadel-Schutzkappe drauf, bis Sie für die Injektion bereit sind. Warten Sie 30 Minuten, damit sich die Fertigspritze auf Raumtemperatur erwärmt, bevor Sie sie benutzen. Dadurch lässt sich das Arzneimittel leichter injizieren. Die Fertigspritze NICHT in die Mikrowelle geben, heisses Wasser darüberlaufen lassen oder direktem Sonnenlicht aussetzen. Die Fertigspritze NICHT einfrieren. Wenn die Fertigspritze eingefroren war, benutzen Sie diese nicht. Die Fertigspritze NICHT schütteln. |
|
1b | Legen Sie alles bereit, was Sie für Ihre Injektion benötigen:
| |
1c |
| Prüfen Sie die Spritze auf äussere Beschädigungen. Kontrollieren Sie das Etikett. Stellen Sie sicher, dass der Name TALTZ auf dem Etikett steht. Stellen Sie auch sicher, dass das Arzneimittel nicht verfallen ist. Die Injektionslösung in der Fertigspritze muss klar sein. Die Farbe reicht von farblos bis leicht gelblich. Wenn Sie Folgendes beobachten, BENUTZEN SIE die Fertigspritze NICHT, und entsorgen Sie sie wie angegeben:
|
1d | Waschen Sie Ihre Hände, bevor Sie sich das Arzneimittel injizieren. | |
1e |
| Wählen Sie Ihre Injektionsstelle. Die Injektion kann in Ihren Bauch (Abdomen), in Ihren Oberschenkel oder in die Rückseite Ihres Arms erfolgen. Die Injektion in Ihren Arm soll von einer anderen Person erledigt werden. Injizieren Sie NICHT in Bereiche, in denen die Haut empfindlich, verletzt, rot oder hart ist, oder an denen Sie Narben oder Dehnungsstreifen haben. Injizieren Sie NICHT in den Bereich innerhalb von 2.5 cm rund um den Nabel. Wechseln Sie Ihre Injektionsstellen. Injizieren Sie NICHT immer exakt in dieselbe Stelle. Zum Beispiel, wenn Ihre letzte Injektion in den linken Oberschenkel erfolgte, soll Ihre nächste Injektion in Ihren rechten Oberschenkel, in Ihren Bauch oder die Rückseite eines Armes gegeben werden. |
Sprechen Sie mit Ihrem Arzt/Apotheker bzw. mit Ihrer Ärztin/Apothekerin oder dem Pflegepersonal, welche Körperstelle am besten für die Injektion Ihrer Dosis geeignet ist. | ||
1f | Bereiten Sie Ihre Haut vor. Reinigen Sie Ihre Haut mit einem Alkoholtupfer. Lassen Sie die Injektionsstelle trocknen, bevor Sie sich Ihr Arzneimittel spritzen. | |



2 | INJEKTION | |||
2a |
| Ziehen Sie die Nadel-Schutzkappe ab und entsorgen Sie sie. Sie dürfen die Nadel-Schutzkappe NICHT wieder aufsetzen - dies könnte die Nadel beschädigen oder Sie könnten sich versehentlich stechen. Berühren Sie NICHT die Nadel. | ||
2b |
| Bilden Sie an der Injektionsstelle behutsam eine Hautfalte und halten Sie sie fest. | ||
Injektion in Bauch, Oberschenkel oder Rückseite | ||||
2c |
| Führen Sie die Nadel in einem Winkel von 45 Grad ein. Lassen Sie nun die Hautfalte behutsam los. Achten Sie darauf, die Nadel an ihrer Position zu halten. | ||
| Lassen Sie die Hautfalte los bevor Sie den Kolben herunterdrücken. | |||
Injektion in Bauch, Oberschenkel oder Rückseite | ||||
2d |
| Drücken Sie den Kolben herunter. Drücken Sie den Kolben langsam und vollständig herunter, bis die Lösung vollständig injiziert ist. Der graue Spritzenkolben muss vollständig bis zum Nadel-Ende der Spritze gedrückt worden sein. Ziehen Sie die Nadel behutsam aus der Haut. Drücken Sie einen Watteball oder ein Stück Gaze auf die Injektionsstelle. Die Injektionsstelle NICHT reiben, da dies blaue Flecken verursachen kann. Es kann sein, dass Sie leicht bluten. Das ist normal. Nach Abschluss der Injektion müssen Sie im Spritzenzylinder die grüne Kolbenstange sehen. | ||
Injektion in Bauch, Oberschenkel oder Rückseite | ||||





3 | ABSCHLUSS | |
3a |
| Entsorgen der Fertigspritze. Setzen Sie die Schutzkappe NICHT wieder auf. Entsorgen Sie die Spritze in ein Nadelbehältnis oder gemäss Anweisung Ihres Arztes/Apothekers bzw. Ärztin/Apothekerin oder des Pflegepersonals. |
Wenn Sie die Spritzen und das Nadelbehältnis entsorgen: | ||
| ||
| ||

Sicherheitshinweise | |
| |
| |
| |
| |
| |
Häufig gestellte Fragen | |
F. | Was mache ich, wenn ich eine Luftblase in der Fertigspritze entdecke? |
A. | Luftblasen in der Fertigspritze sind normal. Taltz wird unter die Haut injiziert (subkutan). Bei dieser Art von Injektion stellen Luftblasen kein Problem dar. Sie sind harmlos und beeinträchtigen nicht Ihre Dosis. |
F. | Was mache ich, wenn sich ein Tropfen Flüssigkeit an der Nadelspitze befindet, nachdem ich die Schutzkappe entfernt habe? |
A. | Es ist in Ordnung, wenn Sie einen Tropfen Flüssigkeit an der Nadelspitze sehen. Das ist harmlos und hat keinen Einfluss auf Ihre Dosis. |
F. | Was mache ich, wenn ich den Kolben nicht herunterdrücken kann? |
A. | Wenn der Kolben verklemmt oder beschädigt ist: |
| |
| |
| |
F. | Woher weiss ich, dass meine Injektion vollständig abgeschlossen ist? |
A. | Wenn Ihre Injektion vollständig abgeschlossen ist: |
| |
| |
Bei Fragen oder für weitere Informationen zu Taltz | |
Es ist wichtig zu wissen, wie Ihr Arzneimittel richtig und sicher injiziert wird. Wenn Sie Fragen zu der Taltz Fertigspritze haben, wenden Sie sich an Ihren Arzt/Apotheker bzw. Ihre Ärztin/Apothekerin oder das Pflegepersonal. | |
Qu'est-ce que Taltz et quand doit-il être utilisé?
Taltz contient le principe actif ixekizumab. L'ixekizumab est un anticorps monoclonal. Les anticorps monoclonaux sont des protéines qui reconnaissent certaines autres protéines du corps humain et se lient à elles.
Taltz appartient à un groupe de médicaments appelé inhibiteurs de l'interleukine (IL). Ce médicament agit en neutralisant l'activité d'une interleukine appelée IL-17A, dont le taux est élevé dans des maladies comme le psoriasis en plaques et l'arthrite psoriasique.
Psoriasis en plaques
Taltz est utilisé sur prescription du médecin pour traiter une affection cutanée appelée «psoriasis en plaques» qui est causée par une inflammation cutanée. Taltz réduit l'inflammation et d'autres symptômes de la maladie tels que la desquamation, les démangeaisons et la douleur.
Taltz est utilisé chez les adultes et chez les enfants et adolescents (dès 6 ans, avec un poids corporel d'au moins 25 kg) atteints d'une forme modérée à sévère de psoriasis en plaques, n'ayant pas répondu ou n'ayant pas toléré d'autres traitements ou chez qui ces traitements sont contre-indiqués.
Arthrite psoriasique
Taltz est indiqué pour traiter une affection appelée «arthrite psoriasique». Il s'agit d'une maladie inflammatoire des articulations, souvent accompagnée d'une maladie de la peau, le psoriasis en plaques.
Taltz réduit les signes et les symptômes de la maladie et améliore la fonction physique (capacité à réaliser les activités quotidiennes habituelles).
Taltz est utilisé sur prescription médicale, seul ou en association avec un autre médicament (antirhumatismal) chez les patients adultes atteints d'arthrite psoriasique, n'ayant pas suffisamment répondu à un traitement conventionnel.
Spondylarthrite ankylosante
Taltz est utilisé pour traiter une affection appelée «spondylarthrite ankylosante». Il s'agit d'une maladie inflammatoire qui affecte principalement la colonne vertébrale et provoque une inflammation des articulations vertébrales.
Taltz est utilisé sur prescription médicale, seul ou en association avec d'autres médicaments pour le traitement de cette maladie, chez les patients adultes atteints de spondylarthrite ankylosante, n'ayant pas suffisamment répondu ou n'ayant pas toléré un traitement conventionnel.
Quand Taltz ne doit-il pas être utilisé?
N'utilisez pas Taltz si vous êtes allergique au principe actif ixekizumab ou à l'un des excipients contenus dans Taltz (voir «Que contient Taltz?»).
Taltz ne doit pas être administré si vous souffrez d'un infection grave, comme p.ex la tuberculose.
Quelles sont les précautions à observer lors de l'utilisation de Taltz?
Adressez-vous à votre médecin ou pharmacien avant d'utiliser Taltz:
- si vous avez actuellement une infection ou si vous avez une infection chronique ou des infections répétées.
- si vous avez la tuberculose.
- si vous souffrez d'une maladie inflammatoire de l'intestin (comme la maladie de Crohn ou la colite ulcéreuse). Veuillez informer immédiatement votre médecin si vous avez des douleurs abdominales, des crampes abdominales, de la diarrhée, une perte de poids ou du sang dans les selles.
- si vous avez été récemment vacciné(e) ou si vous prévoyez de vous faire vacciner pendant le traitement avec Taltz.
- si vous suivez un autre traitement contre le psoriasis en plaques ou l'arthrite psoriasique (médicament ou photothérapie aux rayons ultraviolets).
Taltz peut potentiellement provoquer des effets indésirables graves incluant des infections et des réactions allergiques. Pendant le traitement avec Taltz, vous devez rester attentif à l'apparition de symptômes évoquant ces affections. En cas d'apparition des signes d'infection grave ou de réaction allergique ci-dessous, vous devez arrêter immédiatement d'utiliser Taltz et informer votre médecin ou solliciter une aide médicale.
Signes d'une infection potentiellement grave: fièvre, symptômes grippaux, sueurs nocturnes, sensation de fatigue ou essoufflement; toux qui ne passe pas, chaleur, rougeurs et douleurs cutanées ou éruption cutanée douloureuse avec des vésicules, sensation de brûlure quand vous urinez.
Signes d'une réaction allergique: difficultés à respirer ou à avaler, tension artérielle faible pouvant provoquer des vertiges ou des évanouissements, gonflement du visage, des lèvres, de la bouche ou du cou.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose de 80 mg, c.-à-d. qu'il est essentiellement «sans sodium».
Veuillez informer votre médecin ou votre pharmacien si vous souffrez d'une autre maladie, vous êtes allergique ou vous prenez/utilisez déjà d'autres médicaments (même en automédication !).
Taltz peut-il être utilisé pendant la grossesse ou l'allaitement?
Il est préférable d'éviter l'utilisation de Taltz pendant la grossesse. Les effets de ce médicament chez la femme enceinte ne sont pas connus. Si vous êtes enceinte, si vous pensez être enceinte ou planifiez une grossesse, parlez à votre médecin avant de prendre ce médicament. Pendant le traitement avec Taltz et pendant au moins les 10 semaines suivant la dernière administration de Taltz, vous devez éviter de tomber enceinte en utilisant une méthode de contraception adéquate.
On ne sait pas si le principe actif de Taltz passe dans le lait maternel. En raison de possibles préjudices pour l'enfant allaité, il est recommandé de ne pas allaiter pendant le traitement avec Taltz et pendant au-moins 10 semaines après la dernière dose. Si vous allaitez ou si vous prévoyez d'allaiter, consultez votre médecin avant de prendre ce médicament. Vous et votre médecin déciderez s'il vaut mieux allaiter ou utiliser Taltz. Vous ne devez pas faire les deux simultanément.
Comment utiliser Taltz?
Utilisez toujours ce médicament en suivant exactement les indications de votre médecin, de votre pharmacien ou du personnel soignant. Demandez à votre médecin, votre pharmacien ou au personnel soignant si vous n'êtes pas tout-à-fait certain de comment vous devez utiliser le médicament.
Taltz est administré par injection sous la peau (injection sous-cutanée). Vous et votre médecin déciderez si vous pouvez vous administrer Taltz vous-même.
Il est important de ne pas essayer de vous injecter vous-même ce médicament si vous n'avez pas été formé par votre médecin, votre pharmacien ou le personnel soignant. Un soignant peut également vous administrer Taltz après avoir suivi une formation adaptée.
Lisez soigneusement le manuel d'utilisation de la seringue préremplie avant d'utiliser Taltz.
Quelle est la quantité de Taltz à administrer et quelle est la durée du traitement ?
Chaque seringue préremplie est à usage unique et contient une dose de Taltz de 80 mg. Toute la quantité contenue dans la seringue préremplie dot être injectée.
Psoriasis en plaques chez les adultes
La première dose est de 160 mg (deux injections de 80 mg) administrée par injection sous-cutanée. Ces deux injections vous seront administrées par votre médecin ou le personnel soignant.
Après la première dose, vous recevrez une dose de 80 mg (une injection) aux semaines 2, 4, 6, 8, 10 et 12 et ensuite une dose de 80 mg (une injection) toutes les 4 semaines.
Si vous pesez moins de 100 kg, votre médecin pourra décider d'un dosage alternatif et vous administrer une dose de 80 mg toutes les 4 semaines à partir de la semaine 2.
Psoriasis en plaques chez les enfants et les adolescents (dès 6 ans)
Enfants et adolescents dès 6 ans pesant plus de 50 kg:
Au début du traitement, une dose initiale unique de 160 mg (deux injections de 80 mg) est administrée en injection sous-cutanée. Dès la 4ème semaine après le premier traitement et ensuite toutes les 4 semaines, une dose de 80 mg (une injection de 80 mg) est administrée.
Enfants et adolescents dès 6 ans pesant entre 25 kg et 50 kg:
Au début du traitement, une dose initiale unique de 80 mg (une injection de 80 mg) est administrée en injection sous-cutanée. Dès la 4ème semaine après le premier traitement et ensuite toutes les 4 semaines, une dose de 40 mg est administrée.
Taltz est administré à votre enfant par le médecin ou le personnel soignant.
Pour les doses de 80 mg, toute la quantité de la seringue préremplie doit être administrée.
Les doses de 40 mg ne sont pas disponibles sous forme de seringue péremplie et doivent être préparées par un professionnel de santé qualifié (voir «À quoi faut-il encore faire attention ?»).
Arthrite psoriasique
La première dose est de 160 mg (deux injections de 80 mg) administrée par injection sous-cutanée. Ces deux injections vous seront administrées par votre médecin ou le personnel soignant.
Après la première dose, vous recevrez une dose de 80 mg (une injection) toutes les 4 semaines.
Si vous souffrez à la fois d'arthrite psoriasique et de psoriasis en plaques modéré à sévère, le médecin vous prescrira le dosage pour le psoriasis en plaques.
Spondylarthrite ankylosante
La dose recommandée est de 80 mg (une injection) en injection sous-cutanée toutes les 4 semaines. Pendant le traitement par Taltz, votre médecin peut continuer à vous prescrire d'autres anti-rhumatismaux modificateurs de la maladie conventionnels tels que la sulfasalazine, les corticostéroïdes, les anti-inflammatoires non stéroïdiens et les analgésiques.
Taltz est un traitement au long cours. Votre médecin surveillera régulièrement l'évolution de votre maladie pour vérifier que le traitement produit l'effet attendu.
Si vous avez utilisé plus de Taltz que vous n'auriez dû
Si vous avez par inadvertance reçu trop de Taltz ou si la dose a été reçue plus tôt que prescrit par votre médecin, prévenez-votre médecin.
Si vous avez oublié d'utiliser Taltz
Si vous avez oublié de vous injecter une dose de Taltz, rattrapez-la dès que vous y pensez. Demandez ensuite à votre médecin quand vous devez vous injecter la prochaine dose.
Enfants et adolescents
L'expérience chez les enfants et adolescents dès 6 ans atteints de psoriasis en plaques est limitée au-delà d'un an de traitement. Taltz n'est pas recommandé chez l'enfant et l'adolescent de moins de 6 ans dans le traitement du psoriasis en plaques parce que l'utilisation dans ce groupe d'âge n'a pas été étudiée.
Taltz n'est pas recommandé chez l'enfant et l'adolescent de moins de 18 ans dans le traitement de l'arthrite psoriasique et de la spondylarthrite ankylosante parce que l'utilisation dans ce groupe d'âge n'a pas été étudiée.
Veuillez vous conformer au dosage figurant sur la notice d'emballage ou prescrit par votre médecin. Si vous estimez que l'efficacité du médicament est trop faible ou au contraire trop forte, veuillez vous adresser à votre médecin ou à votre pharmacien.
Quels effets secondaires Taltz peut-il provoquer?
Effets secondaires graves
Arrêtez d'utiliser Taltz et informez immédiatement votre médecin ou sollicitez une aide médicale si vous remarquez l'un des effets indésirables suivants. Votre médecin décidera si et quand vous pouvez reprendre le traitement:
Infection potentiellement grave: les signes peuvent inclure:
- fièvre, symptômes pseudo-grippaux, sueurs nocturnes;
- sensation de fatigue ou essoufflement, toux persistante;
- peau chaude, rouge et douloureuse, ou éruption cutanée douloureuse avec cloques.
Réaction allergique grave: les signes peuvent inclure:
- difficultés à respirer ou à avaler;
- gonflement du visage, des lèvres, de la langue ou de la gorge;
- démangeaisons sévères de la peau avec une éruption cutanée rouge ou des boutons (urticaire).
Les effets secondaires suivants peuvent survenir pendant l'utilisation de Taltz. La plupart sont d'intensité légère à modérée.
Très fréquent (concerne plus d'un utilisateur sur 10)
Infections des voies respiratoires supérieures accompagnées de symptômes tels que mal de gorge et nez bouché (rhinopharyngite), réactions au site d'injection (p.ex. rougeur, douleur).
Fréquent (concerne 1 à 10 utilisateurs sur 100)
Nausées, diarrhée, mycose cutanée, douleur à l'arrière de la gorge.
Occasionnel (concerne 1 à 10 utilisateurs sur 1000)
Mycose buccale, grippe, écoulement nasal, urticaire, sécrétion oculaire avec démangeaisons, rougeur et gonflement (conjonctivite), signes évocateurs d'un faible taux de globules blancs (neutropénie) comme fièvre, maux de gorge ou aphtes dus à des infections, faible taux de plaquettes (thrombocytopénie), maladie inflammatoire de l'intestin (maladie de Crohn, colite ulcéreuse, voir «Quelles sont les précautions à observer lors de l'utilisation de Taltz?»).
Rare (concerne 1 à 10 utilisateurs sur 10 000)
Mycose dans l'œsophage, réaction anaphylactique (allergique) avec des signes tels que gonflement du visage, des lèvres, de la langue ou de la gorge et urticaire (voir «Réaction allergique grave» ci-dessus).
Les effets secondaires rapportés chez les enfants et les adolescents sont globalement similaires à ceux des adultes.
Grippe, urticaire, sécrétion oculaire avec démangeaisons, rougeur et gonflement (conjonctivite), signes évocateurs d'un faible taux de globules blancs (neutropénie) comme fièvre, maux de gorge ou aphtes dus à des infections et réactions d'hypersensibilité (voir «Effets secondaires graves», «Réaction allergique grave» ci-dessus) ont cependant été rapportés fréquemment.
Si vous remarquez des effets secondaires, veuillez en informer votre médecin ou votre pharmacien. Ceci vaut en particulier pour les effets secondaires non mentionnés dans cette notice d'emballage.
À quoi faut-il encore faire attention?
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques concernant le stockage
Stocker au réfrigérateur (2–8 °C). Conserver dans l'emballage original, afin de protéger le contenu de la lumière.
Ne pas congeler. Taltz ne doit pas être utilisé s'il a été congelé.
Stockage temporaire : lorsque la réfrigération n'est pas possible, vous pouvez conserver votre seringue préremplie jusqu'à un total de 5 jours à une température inférieure à 30°C.
Conserver hors de portée des enfants.
Remarques complémentaires
Chaque seringue préremplie est à usage unique.
Ne pas agiter.
Ne pas utiliser si la solution contient des particules ou si elle est trouble et/ou de couleur marron.
Le manuel d'utilisation de la seringue préremplie doit être suivi scrupuleusement.
Pour de plus amples renseignements, consultez votre médecin ou votre pharmacien, qui disposent d'une information détaillée destinée aux professionnels.
Les doses de Taltz 40 mg pour les patients pédiatriques de 25 kg à 50 kg ne peuvent être préparées que par des professionnels de santé conformément à l'information professionnelle.
Que contient Taltz?
Principes actifs
Chaque seringue préremplie préremplie contient 80 mg d'ixekizumab comme principe actif dans 1 ml de solution pour injection.
Excipients
Citrate de sodium dihydraté, acide citrique, chlorure de sodium, polysorbate 80, eau pour préparation injectable.
Numéro d'autorisation
65906 (Swissmedic).
Où obtenez-vous Taltz? Quels sont les emballages à disposition sur le marché?
En pharmacie, sur ordonnance médicale.
Emballages de 1 et 2 seringues préremplies.
Titulaire de l'autorisation
Eli Lilly (Suisse) SA, 1214 Vernier/GE.
Cette notice d'emballage a été vérifiée pour la dernière fois en juillet 2020 par l'autorité de contrôle des médicaments (Swissmedic).
Manuel d’utilisation
Taltz® Ixekizumab 80mg Solution injectable en seringue préremplie |
|
Veuillez lire la notice d’emballage (information destinée aux patients) et ce manuel d’utilisation COMPLETEMENT avant d’utiliser votre seringue préremplie Taltz. |
AVANT D'UTILISER VOTRE SERINGUE PREREMPLIE: INFORMATIONS IMPORTANTES |
|
|

Description de la seringue préremplie |
|

1 | PREPARATION | |
1a | Sortez la seringue préremplie du réfrigérateur. Laissez le capuchon de l’aiguille jusqu’à ce que vous soyez prêt pour l’injection. Attendez 30 minutes pour laisser la seringue préremplie se réchauffer à température ambiante avant de l'utiliser. Cela rend le médicament plus facile à injecter. NE mettez PAS la seringue préremplie au micro-ondes, sous l’eau chaude ou à la lumière directe du soleil. NE PAS congeler la seringue préremplie. Si la seringue préremplie a été congelée, ne l’utilisez pas. NE PAS agiter la seringue préremplie. |
|
1b | Rassemblez les éléments nécessaires à l'injection:
| |
1c |
| Vérifiez que l‘extérieur de la seringue n'est pas abîmé. Vérifiez l'étiquette. Assurez-vous que le nom TALTZ figure sur l'étiquette. Assurez-vous également que le médicament n’est pas périmé. La solution injectable à l'intérieur de la seringue préremplie doit être limpide. Sa couleur peut varier d’incolore à légèrement jaune. Dans les cas suivants, N'UTILISEZ PAS la seringue préremplie et jetez-la selon les recommandations:
|
1d | Lavez-vous les mains avant de procéder à l'injection du médicament. | |
1e |
| Choisissez le site d'injection. Vous pouvez effectuer l'injection dans le ventre (abdomen), la cuisse ou l'arrière de votre bras. L'injection dans le bras doit être effectuée par une autre personne. N'injectez PAS dans des zones où la peau est sensible, blessée, rouge ou dure, ou présente des cicatrices ou des vergetures. Ne faites PAS l’injection à moins de 2,5 cm autour du nombril. Alternez les sites d'injection. N'injectez PAS le produit toujours exactement au même endroit. Par exemple, si votre dernière injection a été faite dans la cuisse gauche, vous pouvez faire l'injection suivante dans la cuisse droite, le ventre ou à l'arrière de l'un des deux bras. |
Discutez avec votre médecin, votre pharmacien ou le personnel soignant de quelle partie du corps est la plus appropriée pour l’injection de votre dose. | ||
1f | Préparez votre peau. Nettoyez votre peau avec une compresse imbibée d'alcool. Laissez sécher le site d'injection avant de procéder à l'injection du médicament. | |



2 | INJECTION | ||
2a |
| Retirez le capuchon de l'aiguille et jetez-le. NE remettez PAS le capuchon sur l'aiguille - vous risqueriez d'abîmer l'aiguille ou de vous blesser accidentellement. NE touchez PAS l'aiguille. | |
2b |
| Pincez doucement la peau et maintenez un pli de peau dans lequel l'injection sera effectuée. | |
Injection dans le ventre, la cuisse ou l’arrière du bras | |||
2c |
| Insérez l'aiguille en l'inclinant de 45 degrés. Ensuite lâchez doucement le pli de peau. Maintenez bien l'aiguille en place. | |
| Lâchez le pli de peau avant de pousser sur le piston. | ||
Injection dans le ventre, la cuisse ou l’arrière du bras | |||
2d |
| Poussez sur le piston. Poussez lentement et complètement sur le piston jusqu'à ce que la totalité de la solution ait été injectée. Le piston gris de la seringue doit être poussé jusqu’au bout vers l’aiguille. Retirez doucement l'aiguille de votre peau. Appuyez avec un morceau de coton ou de gaze sur le site d'injection. NE frottez PAS le site d'injection pour ne pas provoquer d'ecchymoses. Il peut y avoir un léger saignement. C'est normal. Vous devez voir la tige du piston verte dans le corps de la seringue lorsque l'injection est terminée. | |
Injection dans le ventre, la cuisse ou l’arrière du bras | |||





3 | FIN | |
3a |
| Elimination de la seringue préremplie. NE remettez PAS le capuchon sur l'aiguille. Jetez la seringue dans un conteneur à aiguilles ou selon les recommandations de votre médecin, de votre pharmacien ou du personnel soignant. |

Lorsque vous jetez la seringue et le conteneur à aiguilles: | |
| |
| |
Conseils de sécurité | |
| |
| |
| |
| |
| |
Foire aux questions | |
Q. | Que dois-je faire si je vois des bulles d'air dans la seringue préremplie? |
R. | Il est normal que la seringue préremplie contienne des bulles d'air. Taltz est injecté sous votre peau (injection sous-cutanée). La présence de bulles d'air ne pose pas de problème pour ce type d'injection. Cela n'est pas dangereux et n'a pas d'effet sur votre dose. |
Q. | Que faire s’il y a une goutte de liquide au bout de l'aiguille quand je retire le capuchon? |
R. | Il est normal de voir une goutte de liquide au bout de l'aiguille. Cela n'est pas dangereux et n'a pas d'effet sur votre dose. |
Q. | Que faire si je n'arrive pas à pousser sur le piston? |
R. | Si le piston est coincé ou abîmé: |
| |
| |
Q. | Comment savoir si l'injection est terminée? |
R. | Lorsque l'injection est terminée: |
| |
| |
Si vous avez des questions ou pour de plus amples informations sur Taltz | |
Il est important de savoir comment injecter votre médicament de manière correcte et en toute sécurité. Si vous avez des questions sur la seringue préremplie Taltz, adressez-vous à votre médecin, votre pharmacien ou au personnel soignant. | |
Che cos'è Taltz e quando si usa?
Taltz contiene il principio attivo Ixekizumab. Ixekizumab è un anticorpo monoclonale. Gli anticorpi monoclonali sono proteine che riconoscono altre specifiche proteine nell'organismo e si legano ad esse.
Taltz appartiene a un gruppo di farmaci definiti inibitori dell'interleuchina (inibitori IL). Questo medicamento agisce neutralizzando l'attività di un'interleuchina denominata IL-17A, che si presenta in concentrazioni elevate in caso di patologie quali la psoriasi a placche e artrite psoriasica.
Psoriasi a placche
Taltz è utilizzato dietro prescrizione medica per il trattamento di una malattia della pelle definita «psoriasi a placche» provocata da un'infiammazione che colpisce la pelle. L'uso di Taltz allevia i sintomi della malattia quali desquamazione, prurito e dolore.
Taltz è usato negli adulti e nei bambini e adolescenti (dai 6 anni, con un peso corporeo di almeno 25 kg) affetti da psoriasi a placche di grado da moderato a grave, che non hanno reagito ad altre terapie, nei quali queste terapie sono controindicate o che non tollerano queste terapie.
Artrite psoriasica
Taltz è utilizzato per il trattamento di una malattia denominata «artrite psoriasica». Si tratta di una malattia infiammatoria delle articolazioni, spesso accompagnata da una malattia della pelle, la psoriasi a placche.
Taltz riduce i segni e i sintomi della malattia e migliora la funzione fisica (capacità di svolgere le normali attività quotidiane).
Taltz è usato da solo o in associazione a un altro medicamento (antireumatico) su prescrizione del medico in pazienti adulti con artrite psoriasica che non hanno risposto in modo sufficiente alla terapia convenzionale.
Spondilite anchilosante
Taltz è utilizzato per il trattamento di una malattia denominata «spondilite anchilosante». Si tratta di una malattia infiammatoria che colpisce principalmente la colonna vertebrale e provoca un'infiammazione delle articolazioni della colonna vertebrale.
Taltz è usato da solo o in associazione ad altri medicamenti su prescrizione del medico per trattare questa malattia in pazienti adulti con spondilite anchilosante, che non hanno risposto in modo sufficiente alla terapia convenzionale o non l'hanno tollerata.
Quando non si può usare Taltz?
Taltz non va usato in caso di allergia al principio attivo ixekizumab o a uno qualsiasi degli eccipienti (vedere «Cosa contiene Taltz?»).
Taltz non deve essere somministrato nel caso in cui il paziente sia affetto da una grave infezione, per esempio la tubercolosi.
Quando è richiesta prudenza nell'uso di Taltz?
Informi il suo medico o il suo farmacista prima di assumere Taltz nel caso in cui:
- soffre attualmente di un'infezione o soffre di un'infezione cronica o di infezioni ripetute.
- soffre di tubercolosi.
- soffre di una malattia infiammatoria dell'intestino (come il morbo di Crohn o la colite ulcerosa). Informi immediatamente il suo medico se ha mal di stomaco, crampi addominali, diarrea, perdita di peso o sangue nelle feci.
- è stato/a recentemente vaccinato/a o prevede di esserlo durante il trattamento con Taltz.
- sta seguendo un'altra cura per la psoriasi a placche o artrite psoriasica (farmacologica o fototerapia a base di radiazioni ultraviolette (UV)).
Taltz può provocare effetti collaterali potenzialmente gravi, tra cui infezioni e reazioni allergiche. Durante la terapia con Taltz occorre prestare attenzione agli eventuali sintomi di tali patologie. Se si riscontrano i seguenti sintomi, indicativi di un'infezione potenzialmente grave o di una reazione allergica, interrompere subito l'assunzione di Taltz e informare il proprio medico o richiedere assistenza medica.
Sintomi di infezione potenzialmente grave: febbre, sintomi simil-influenzali, sudorazione notturna, senso di stanchezza o affanno, tosse persistente, cute calda, arrossata e dolente o eruzione cutanea dolente con formazione di vescicole, bruciore durante la minzione.
Sintomi di una reazione allergica: difficoltà a respirare o deglutire, ipotensione che può provocare vertigini o intontimento, gonfiore del viso, delle labbra, della bocca o della gola.
Questo medicamento contiene meno di 1 mmol (23 mg) di sodio per dose di 80 mg, cioè è essenzialmente «senza sodio».
Informi il suo medico o il suo farmacista nel caso in cui soffra di altre malattie, soffra di allergie o assuma o applichi esternamente altri medicamenti (anche se acquistati di sua iniziativa!).
Si può usare Taltz durante la gravidanza o l'allattamento?
L'uso di Taltz in gravidanza dovrebbe essere evitato. Gli effetti del medicamento sulle donne in gravidanza non sono noti. Se è in corso una gravidanza, se sospetta o sta pianificando una gravidanza, chieda consiglio al medico. Durante il trattamento con Taltz e per almeno 10 settimane dall'ultima assunzione di Taltz occorre evitare una gravidanza usando idonei mezzi contraccettivi.
Non è noto se il principio attivo di Taltz sia escreto nel latte materno. È sconsigliato allattare durante il trattamento con Talz e per almeno 10 settimane dopo la fine dell'ultima dose per evitare eventuali danni al lattante. Prima di iniziare ad assumere Taltz chiedere consiglio al medico nel caso si stia allattando o si intenda allattare. La paziente deve decidere con il medico se allattare oppure assumere Taltz. Non è possibile fare entrambe le cose.
Come usare Taltz?
Usare sempre Taltz attenendosi scrupolosamente alle indicazioni del medico, del farmacista o dell'operatore sanitario. Se non si è sicuri di come utilizzare il medicamento, rivolgersi al medico, al farmacista o all'operatore sanitario.
Taltz è iniettato sotto cute (per via sottocutanea). Il paziente deve decidere con il medico se iniettarsi Taltz da solo.
È importante non provare a praticare l'iniezione da soli prima che il medico, il farmacista o l'operatore sanitario abbiano mostrato come si fa. Anche un assistente familiare può eseguire l'iniezione di Taltz dopo essere stato adeguatamente preparato.
Leggere attentamente le Istruzioni per l'uso della siringa preriempita prima di utilizzare Taltz.
Che dose di Taltz si deve assumere e per quanto tempo?
Le siringhe preriempite sono monouso e rilasciano una dose di Taltz di 80 mg. Tutta la dose contenuta nella siringa preriempita deve essere iniettata.
Psoriasi a placche negli adulti
La prima volta si somministrano 160 mg (due iniezioni da 80 mg) di medicamento con due iniezioni sottocutanee. Queste prime iniezioni sono praticate dal medico o dall'operatore sanitario.
Dopo questa prima somministrazione il paziente riceve dosi da 80 mg (un'iniezione) nelle settimane 2, 4, 6, 8, 10 e 12 e poi una dose di 80 mg (un'iniezione) ogni 4 settimane.
Se pesa meno di 100 kg, il suo medico deciderà se somministrarle un dosaggio alternativo e a partire dalla settimana 2 le somministrerà la dose di 80 mg tutte le 4 settimane.
Psoriasi a placche nei bambini e adolescenti (dai 6 anni)
Bambini e adolescenti dai 6 anni con un peso corporeo superiore a 50 kg:
All'inizio del trattamento viene somministrata una singola dose iniziale di 160 mg (due iniezioni da 80 mg) per via sottocutanea. Dalla quarta settimana dopo il primo trattamento e successivamente viene somministrata una dose di 80 mg (una iniezione da 80 mg) ogni 4 settimane.
Bambini e adolescenti dai 6 anni con un peso corporeo da 25 kg a 50 kg:
All'inizio del trattamento viene somministrata una singola dose iniziale di 80 mg (una iniezione da 80 mg) per via sottocutanea. Dalla quarta settimana dopo il primo trattamento e successivamente viene somministrata una dose di 40 mg ogni 4 settimane.
Il medico o l'operatore sanitario somministrerà Taltz al suo bambino.
Per la dose di 80 mg, deve essere somministrato tutto il contenuto della siringa preriempita.
Le dosi da 40 mg non sono diponibili sotto forma di siringa preriempita e devono essere preparate da un professionista sanitario qualificato (vedere «Di che altro occorre tener conto?»).
Artrite psoriasica
La prima volta si somministrano 160 mg (due iniezioni da 80 mg) di medicamento con due iniezioni sottocutanee. Queste prime iniezioni sono praticate dal medico o dall'operatore sanitario.
Dopo questa prima somministrazione il paziente riceve dosi da 80 mg (un'iniezione) ogni 4 settimane.
Se, oltre all'artrite psoriasica, soffre anche di psoriasi a placche da moderata a grave, il suo medico le prescriverà il dosaggio per la psoriasi a placche.
Spondilite anchilosante
La dose raccomandata è di 80 mg (un'iniezione) somministrata per via sottocutanea ogni 4 settimane.
Durante il trattamento con Taltz, il medico può continuare se necessario a prescrivere altri farmaci antireumatici convenzionali modificanti la malattia, come sulfasalazina, corticosteroidi, farmaci antinfiammatori non steroidei e analgesici.
Taltz è destinato all'uso nelle terapie a lungo termine. Il medico controllerà regolarmente il decorso della malattia per verificare che la cura produca l'effetto auspicato.
In caso di assunzione di una dose di Taltz superiore a quella prescritta
In caso di assunzione accidentale di una dose di Taltz maggiore di quella prescritta o di assunzione anticipata, informare il medico.
Nel caso si dimentichi di assumere Taltz
Nel caso si dimentichi di iniettare una dose di Taltz, farlo non appena si ricorda. Poi chiedere al medico quando assumere la dose successiva.
Bambini e adolescenti
L'esperienza nei bambini e adolescenti con psoriasi a placche dai 6 anni è limitata alla durata della terapia di un anno. L'uso di Taltz non è consigliato nei bambini e adolescenti di età inferiore a 6 anni per il trattamento della psoriasi a placche, in quanto l'uso in questa fascia d'età non è stato testato.
L'uso di Taltz non è consigliato nei bambini e adolescenti di età inferiore a 18 anni per il trattamento dell'artrite psoriasica e della spondilite anchilosante in quanto l'uso in questa fascia di età non è stato testato.
Si attenga alla posologia indicata nel foglietto illustrativo o prescritta dal suo medico. Se ritiene che l'azione del medicamento sia troppo debole o troppo forte ne parli al suo medico o al suo farmacista.
Quali effetti collaterali può avere Taltz?
Effetti collaterali gravi
Interrompere l'assunzione di Taltz e consultare immediatamente un medico o richiedere assistenza medica nel caso si osservi uno dei seguenti effetti collaterali. Il medico deciderà se e quando riprendere la cura.
Infezione potenzialmente grave: Sintomi che possono presentarsi:
- febbre, sintomi simil-influenzali, sudorazione notturna;
- senso di stanchezza o affanno; tosse persistente;
- cute calda, arrossata e dolente o eruzione cutanea dolente con formazione di vescicole.
Grave reazione allergica: Sintomi che possono presentarsi:
- difficoltà a respirare o deglutire;
- gonfiore del viso, delle labbra, della lingua o della gola;
- prurito intenso alla cute con eruzione e arrossamento o pomfi in rilievo (orticaria).
I seguenti effetti collateralipossono verificarsi durante l'uso di Taltz. La maggior parte presenta una gravità di grado lieve o moderato.
Molto comune (riguarda più di 1 utilizzatore su 10)
Infezioni delle vie respiratorie superiori con sintomi quali mal di gola e nasochiuso (rinofaringite), reazioni nel sito di iniezione (ad es. pelle arrossata, dolore).
Comune (riguarda da 1 a 10 utilizzatori su 100)
Nausea, diarrea, micosi della pelle, dolore alla parte posteriore della gola.
Non comune (riguarda da 1 a 10 utilizzatori su 1000)
Micosi del cavo orale, influenza, naso che gocciola, orticaria, lacrimazione accompagnata da prurito, arrossamento e gonfiore (congiuntivite), segnali di diminuzione dei globuli bianchi (neutropenia) quali febbre, mal di gola o ulcere del cavo orale causate da infezioni, valori bassi delle piastrine nel sangue bassa (trombocitopenia), malattia infiammatoria dell'intestino (morbo di Chron, colite ulcerosa, vedere «Quando è richiesta prudenza nell'uso di Taltz?»).
Raro (riguarda da 1 a 10 utilizzatori su 10 000)
Candidosi esofagea, reazione anafilattica (allergica) con segni quali gonfiore del viso, delle labbra, della lingua o del collo e orticaria (vedi sopra «Grave reazione allergica»).
Gli effetti collaterali segnalati nei bambini e adolescenti sono nel complesso paragonabili a quelli segnalati negli adulti.
Tuttavia, sono state riportate spesso influenza, orticaria, lacrimazione accompagnata da prurito, arrossamento e gonfiore (congiuntivite), segnali di diminuzione dei globuli bianchi (neutropenia) quali febbre, mal di gola o ulcere della bocca causate da infezioni e reazioni di ipersensibilità (vedere «Effetti collaterali gravi», «Grave reazione allergica» sopra).
Se osserva effetti collaterali, si rivolga al suo medico o farmacista, soprattutto se si tratta di effetti collaterali non descritti in questo foglietto illustrativo.
Di che altro occorre tener conto?
Il medicamento non dev'essere utilizzato oltre la data indicata con «EXP» sul contenitore.
Indicazione di stoccaggio
Conservare in frigorifero (2-8°C). Conservare nella confezione originale per tenere il contenuto al riparo dalla luce.
Non congelare. Non utilizzare Taltz che sia stato congelato.
Conservazione temporanea: quando non è possibile conservare la siringa preriempita in frigorifero, è possibile conservarla a temperatura inferiore a 30 °C fino ad un massimo di 5 giorni.
Conservare fuori dalla portata dei bambini.
Ulteriori indicazioni
Ogni siringa preriempita è monouso.
Non agitare.
Non utilizzare se si osservano particelle o se la soluzione è torbida e/o chiaramente marrone.
Le istruzioni per l'uso della siringa preriempita devono essere osservate scrupolosamente.
Il medico o il farmacista, che sono in possesso di un'informazione professionale dettagliata, possono darle ulteriori informazioni.
Le dosi da 40 mg di Taltz per pazienti pediatrici da 25 kg a 50 kg di peso possono essere preparate esclusivamente dal personale sanitario in conformità all'informazione professionale.
Cosa contiene Taltz?
Principi attivi
Ogni siringa preriempita contiene 80 mg di principio attivo Ixekizumab in 1 ml di soluzione iniettabile.
Sostanze ausiliarie
Citrato di sodio diidrato, acido citrico, cloruro di sodio, polisorbato 80, acqua per iniezione.
Numero dell'omologazione
65906 (Swissmedic).
Dove è ottenibile Taltz? Quali confezioni sono disponibili?
In farmacia, dietro presentazione della prescrizione medica.
Confezioni da 1 e 2 siringhe preriempite.
Titolare dell'omologazione
Eli Lilly (Suisse) SA, 1214 Vernier/GE.
Questo foglietto illustrativo è stato controllato l'ultima volta nel luglio 2020 dall'autorità competente in materia di medicamenti (Swissmedic).
Istruzioni per l'uso
Taltz® Ixekizumab 80 mg Soluzione iniettabile in siringa preriempita |
|
Legga attentamente e PER INTERO il foglietto illustrativo (Informazione destinata ai pazienti) e le presenti istruzioni prima di usare la siringa preriempita Taltz. |
PRIMA DI USARE LA SIRINGA PRERIEMPITA: COSE IMPORTANTI DA SAPERE |
|
|

Descrizione della siringa preriempita |
|

1 | PRIMA DI COMINCIARE | |
1a | Prendere la siringa preriempita dal frigorifero. Lasci il cappuccio dell’ago finché non è pronto per fare l’iniezione. Aspettare 30 minuti per consentire che la siringa raggiunga la temperatura ambiente prima di usarla. In questo modo è più semplice iniettare il medicamento. NON inserire la siringa preriempita nel forno a microonde, non versarci sopra acqua calda o esporla alla luce diretta del sole. NON congelare la siringa preriempita. Se la siringa preriempita è stata congelata, non utilizzarla. NON agitare la siringa preriempita. |
|
1b | Predisporre tutto ciò che serve per l'iniezione:
| |
1c |
| Verificare che la siringa non presenti difetti esteriori. Controllare l'etichetta. Accertarsi che l'etichetta riporti la denominazione TALTZ. Accertarsi anche che il medicamento non sia scaduto. La soluzione iniettabile contenuta nella siringa preriempita deve essere limpida. La soluzione deve essere da incolore a leggermente gialla. Se si riscontra una delle seguenti situazioni, NON UTILIZZARE la siringa preriempita e smaltirla come riportato:
|
1d | Lavarsi le mani prima di iniettare il medicamento. | |
1e |
| Scegliere il sito di iniezione. L'iniezione può essere praticata nell'addome, nella coscia o nella parte posteriore del braccio. L'iniezione nel braccio deve essere eseguita da un'altra persona. NON praticare l'iniezione in zone dove la cute è sensibile, lesionata, arrossata o indurita oppure dove sono presenti cicatrici o smagliature. NON praticare l'iniezione nel raggio di 2,5 cm dall'ombelico. Alternare i siti di iniezione. NON praticare sempre l'iniezione esattamente nello stesso punto. Ad esempio, se l’ultima iniezione è stata fatta nella coscia sinistra, l’iniezione successiva deve essere effettuata nella coscia destra, nell’addome o nella parte posteriore di un braccio. |
Scegliere con il medico, il farmacista o un operatore sanitario la parte del corpo più adatta per iniettare la sua dose | ||
1f | Preparare la sua cute. Detergere la sua cute con un tampone imbevuto d'alcol. Lasciare asciugare il sito di iniezione prima di iniettare il medicamento. | |



2 | INIEZIONE | ||
2a |
| Rimuovere e gettare il cappuccio di protezione dell'ago. NON rimettere il cappuccio dell'ago - facendolo si potrebbe danneggiare l'ago o pungersi accidentalmente. NON toccare l'ago. | |
2b |
| Stringere delicatamente e tenere la porzione di cute dove effettuerà l’iniezione. | |
Iniezione nell’addome, nella coscia o nella parte posteriore del braccio | |||
2c |
| Inserire l'ago con un angolo di 45 gradi. Lasciare andare delicatamente la cute. Assicurarsi di tenere l'ago in posizione. | |
| Lasciare andare la plica di cute prima di spingere lo stantuffo. | ||
Iniezione nell’addome, nella coscia o nella parte posteriore del braccio | |||
2d |
| Premere lo stantuffo. Premere lentamente lo stantuffo fino in fondo, finché non è stato iniettato tutto il medicamento. Lo stantuffo grigio deve essere spinto fino alla fine della siringa. Estrarre delicatamente l'ago dalla cute. Premere un batuffolo di ovatta o una garza sul sito di iniezione. NON strofinare il sito di iniezione perché ciò potrebbe causare la comparsa di lividi. Si può verificare un leggero sanguinamento. Quest'evenienza è da considerarsi normale. Quando l'iniezione è terminata si deve vedere l'asta verde dello stantuffo attraverso il corpo della siringa. | |
Iniezione nell’addome, nella coscia o nella parte posteriore del braccio | |||





3 | CONCLUSIONE | |
3a |
| Smaltimento della siringa preriempita. NON rimettere il cappuccio dell'ago. Gettare la siringa in un contenitore porta-aghi o come indicato dal medico, dal farmacista o dall'operatore sanitario. |
Smaltimento delle siringhe e del contenitore porta-aghi: | ||
| ||
| ||

Avvertenze per la sicurezza | |
| |
| |
| |
| |
| |
Domande più frequenti | |
D. | Cosa fare se si nota una bolla d'aria nella siringa preriempita? |
R. | La presenza di bolle d'aria nella siringa preriempita è normale. Taltz è iniettato sotto cute (per via sottocutanea). In questo tipo di iniezione le bolle d'aria non costituiscono un problema. È una normale evenienza e non influisce sul dosaggio. |
D. | Cosa fare se c'è una goccia di liquido sulla punta dell'ago quando si rimuove il cappuccio di protezione? |
R. | Va bene se si nota una goccia di liquido sulla punta dell'ago. È una normale evenienza e non influisce sul dosaggio. |
D. | Cosa fare se non si riesce a premere lo stantuffo? |
R. | Se lo stantuffo è bloccato o danneggiato: |
| |
| |
| |
D. | Come si capisce che l'iniezione è terminata? |
R. | L'iniezione è terminata quando: |
| |
| |
In caso di domande o per maggiori informazioni su Taltz | |
È importante sapere come iniettare il medicamento correttamente e in sicurezza. Se ha domande sulla siringa preriempita Taltz, si rivolga al suo medico, al suo farmacista o a un operatore sanitario. | |
Zusammensetzung
Wirkstoffe
Ixekizumab (aus gentechnologisch hergestellten CHO (Chinese Hamster Ovary)-Zellen).
Hilfsstoffe
Natriumcitrat-Dihydrat, Citronensäure, Natriumchlorid, Polysorbat 80, Wasser für Injektionszwecke q.s. ad solutionem pro 1 ml.
Gesamtnatriumgehalt: 5.8 mg/ml.
Darreichungsform und Wirkstoffmenge pro Einheit
Taltz 80 mg Injektionslösung in einer Fertigspritze
Jede Fertigspritze enthält 80 mg Ixekizumab in 1 ml (80 mg/ml).
Taltz 80 mg Injektionslösung in einem Fertigpen
Jeder Fertigpen enthält 80 mg Ixekizumab in 1 ml (80 mg/ml).
Indikationen/Anwendungsmöglichkeiten
Plaque-Psoriasis
Taltz ist zur Behandlung erwachsener Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis indiziert, die auf andere systemische Therapien (einschliesslich Ciclosporin oder Methotrexat oder PUVA) nicht angesprochen haben, bei denen diese Therapien kontraindiziert sind oder die diese Therapien nicht tolerieren.
Plaque-Psoriasis bei Kindern und Jugendlichen
Taltz ist zur Behandlung der mittelschweren bis schweren Plaque-Psoriasis bei Kindern und Jugendlichen ab 6 Jahren mit einem Körpergewicht von mindestens 25 kg indiziert, die auf andere systemische Therapien (einschliesslich Ciclosporin oder Methotrexat oder PUVA) nicht angesprochen haben, bei denen diese Therapien kontraindiziert sind oder die diese Therapien nicht tolerieren.
Psoriasis-Arthritis
Taltz, alleine oder in Kombination mit konventionellen krankheitsmodifizierenden Antirheumatika (DMARD, disease-modifying anti-rheumatic drugs), ist zur Behandlung erwachsener Patienten mit aktiver Psoriasis-Arthritis indiziert, die auf eine Behandlung mit einem oder mehreren DMARDs unzureichend angesprochen haben oder diese nicht vertragen haben.
Ankylosierende Spondylitis (Morbus Bechterew)
Taltz ist indiziert zur Behandlung der schweren aktiven ankylosierenden Spondylitis bei erwachsenen Patienten, die auf eine konventionelle Therapie (z.B. nichtsteroidale entzündungshemmende Medikamente [NSAIDs]) unzureichend angesprochen haben oder diese nicht vertragen.
Dosierung/Anwendung
Die Anwendung von Taltz sollte unter Anleitung und Aufsicht eines in Diagnose und Behandlung der für Taltz indizierten Erkrankungen, erfahrenen Arztes erfolgen.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Übliche Dosierung
Plaque-Psoriasis bei Erwachsenen
Die empfohlene Dosis beträgt 160 mg als subkutane Injektion (zwei 80 mg Injektionen) in Woche 0, gefolgt von 80 mg (eine Injektion) in den Wochen 2, 4, 6, 8, 10 und 12, und danach 80 mg (eine Injektion) alle 4 Wochen.
Bei Patienten <100 kg kann ein alternatives Dosisschema mit 160 mg in Woche 0 und ab Woche 2 80 mg alle 4 Wochen erwogen werden (siehe «Eigenschaften/Wirkungen»).
Plaque-Psoriasis bei Kindern und Jugendlichen (ab 6 Jahren)
Die empfohlene Dosis für die subkutane Injektion bei pädiatrischen Patienten ab 6 Jahren mit einem Körpergewicht von mindestens 25 kg basiert auf den folgenden Gewichtsklassen:
Gewicht des pädiatrischen Patienten | Einmalige Initialdosis (Woche 0) | Dosis ab Woche 4 und danach alle 4 Wochen |
|---|---|---|
Über 50 kg | 160 mg (zwei Injektionen zu 80 mg) | 80 mg (eine Injektion zu 80 mg) |
25 bis 50 kg | 80 mg (eine Injektion zu 80 mg) | 40 mg |
Bei pädiatrischen Patienten, bei denen 80 mg verschrieben werden, kann die komplette Dosis direkt aus der Fertigspritze oder aus dem Fertigpen verwendet werden.
Anweisungen zur Vorbereitung einer Injektionsspritze mit Taltz 40 mg Dosen aus einer Fertigspritze mit 80 mg finden Sie unter «Sonstige Hinweise / Hinweise für die Handhabung».
Die Anwendung von Taltz wird bei Kindern mit einem Körpergewicht unter 25 kg nicht empfohlen.
Psoriasis-Arthritis
Die empfohlene Dosis beträgt 160 mg als subkutane Injektion (zwei 80 mg Injektionen) in Woche 0, gefolgt von 80 mg (eine Injektion) alle 4 Wochen.
Für Patienten mit Psoriasis-Arthritis und begleitender mittelschwerer bis schwerer Plaque-Psoriasis sind die Dosierungsempfehlungen dieselben wie für die Plaque-Psoriasis.
Ankylosierende Spondylitis
Die empfohlene Dosis beträgt 80 mg (eine Injektion) als subkutane Injektion alle 4 Wochen.
Bei der ankylosierenden Spondylitis können konventionelle DMARDs (z.B. Sulfasalazin), Kortikosteroide, NSAIDs und/oder Analgetika während der Behandlung mit Taltz angewendet werden.
Therapiedauer
In allen Indikationen (Plaque-Psoriasis, Psoriasis-Arthritis, ankylosierende Spondylitis), sollte bei Patienten, die nach 16 bis 20 Wochen Therapie kein Ansprechen gezeigt haben, ein Abbruch der Therapie in Erwägung gezogen werden. Einige Patienten, die initial ein teilweises Ansprechen gezeigt haben, können später, bei Fortsetzung der Therapie über 20 Wochen hinaus, eine Besserung zeigen.
Patienten mit Leberfunktionsstörungen
Taltz wurde in dieser Patientenpopulation nicht untersucht.
Patienten mit Nierenfunktionsstörungen
Taltz wurde in dieser Patientenpopulation nicht untersucht.
Ältere Patienten
Von den 4204 Patienten mit Plaque Psoriasis, die in klinischen Entwicklungsstudien Taltz erhalten haben, waren insgesamt 301 Patienten 65 Jahre alt oder älter und 36 Patienten 75 Jahre alt oder älter. Von den 1118 Patienten mit Psoriasis-Arthritis, die in klinischen Studien Taltz erhalten haben, waren insgesamt 122 Patienten 65 Jahre alt oder älter und 6 Patienten 75 Jahre alt oder älter. Dosisanpassungen sind nicht erforderlich (siehe «Pharmakokinetik»).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Taltz bei Kindern unter 6 Jahren bei der Behandlung von Plaque-Psoriasis wurden bisher nicht belegt. Die Erfahrung bei Kindern und Jugendlichen ist limitiert. Die Wirksamkeit und Sicherheit bei der Anwendung über ein Jahr hinaus sind ungenügend belegt. Die Sicherheit und Wirksamkeit von Taltz bei Kindern und Jugendlichen unter 18 Jahren bei der Behandlung von Psoriasis-Arthritis und ankylosierenden Spondylitis wurden bisher nicht belegt.
Art der Anwendung
Vor Therapie-Beginn muss der Arzt resp. die Ärztin Folgendes sicherstellen:
Der Patient, bzw. bei Kindern und Jugendlichen die erziehungsberechtigte Person versteht, dass Taltz eine neuartige Therapie mit limitierter Erfahrung und unbekannten Langzeitrisiken darstellt.
Taltz wird subkutan alternierend am Oberarm, Bauch und Oberschenkel injiziert.
Von Psoriasis befallene Hautbereiche sollen als Injektionsstellen vermieden werden.
Nach sachgemässer Schulung zur subkutanen Injektionstechnik können sich erwachsene Patienten Taltz selbst injizieren, wenn der behandelnde Arzt dies für angebracht hält. Umfangreiche Anweisungen zur Anwendung sind in der Bedienungsanleitung der Spritze und des Pens beschrieben.
Plaque-Psoriasis bei Kindern und Jugendlichen:
Bei pädiatrischen Patienten mit einem Gewicht von mehr als 50 kg können Pflegepersonen nach der Schulung zur subkutanen Injektionstechnik Injektionen verabreichen.
Bei pädiatrischen Patienten von 25 kg bis zu 50 kg müssen Ixekizumab-Dosen von 40 mg von einer qualifizierten medizinischen Fachperson vorbereitet und verabreicht werden. Verwenden Sie nur die im Handel erhältliche Taltz 80 mg/1 ml-Fertigspritze, wenn Sie die vorgeschriebenen pädiatrischen Dosen von 40 mg vorbereiten (siehe «Sonstige Hinweise / Hinweise für die Handhabung»).
Nach der Verabreichung von Taltz sind Kinder und Jugendliche über einen angemessenen Zeitraum medizinisch nachzubeobachten.
Kontraindikationen
Schwere Überempfindlichkeit gegen den Wirkstoff oder einen der Hilfsstoffe.
Schwere aktive Infektionen (z.B. aktive Tuberkulose, Sepsis, schwere opportunistische Infektionen).
Warnhinweise und Vorsichtsmassnahmen
Infektionen
Die Therapie mit Taltz ist dosisabhängig mit einer erhöhten Infektionsrate wie Infektionen der oberen Atemwege, orale Candidiasis, Konjunktivitis oder Tinea-Infektionen assoziiert (siehe «Unerwünschte Wirkungen»). Unter immunomodulatorischer Therapie kann es zur Reaktivierung latenter Infektionen kommen.
Bei Patienten mit chronischer oder aktiver Infektion oder rezidivierenden Infektionen in der Vorgeschichte muss Taltz mit Vorsicht angewendet werden. Taltz sollte nicht an Patienten mit aktiver Infektion, insbesondere HIV-, HBV- oder HCV-Infektionen, verabreicht werden. Wenn sich unter Therapie mit Taltz eine solche Infektion entwickelt, ist eine sorgfältige Überwachung erforderlich. Die Patienten sollten angewiesen werden, bei Auftreten von Anzeichen oder Symptomen einer Infektion ärztlichen Rat einzuholen. Die Therapie mit Taltz muss bei Nicht-Ansprechen einer antiinfektiösen Standardtherapie sowie bei schwerwiegenden Infekten beendet werden. Die Therapie mit Taltz ist erst fortzusetzen, wenn die Infektion abgeklungen ist.
Patienten mit aktiver Tuberkulose dürfen Taltz nicht erhalten. Bei Patienten mit latenter Tuberkulose sollte vor dem Beginn der Taltz Therapie eine antituberkulöse Therapie erwogen werden.
Maligne Erkrankungen
Daten aus klinischen Studien über 1 Jahr zeigten kein erhöhtes Risiko für maligne Erkrankungen. Weitere Untersuchungen zur Langzeitsicherheit sind noch nicht vorhanden. Da es sich bei Psoriasis-Patienten um eine Risikopopulation handelt, sollten Psoriasis-Patienten vor und unter der Behandlung mit Taltz auf das Vorliegen von Hauttumoren untersucht werden.
Überempfindlichkeitsreaktionen
Schwerwiegende Überempfindlichkeitsreaktionen, einschliesslich Fälle von Anaphylaxie, Angioödem und Urtikaria wurden berichtet. Wenn eine schwerwiegende Überempfindlichkeitsreaktion auftritt, muss die Anwendung von Taltz umgehend abgebrochen und eine geeignete Therapie eingeleitet werden.
Depression
Depression ist eine bekannte Komorbidität bei Patienten mit ankylosierender Spondylitis. Bei Patienten mit ankylosierender Spondylitis mit Depressionsrisiko ist Vorsicht geboten.
Entzündliche Darmerkrankungen, einschliesslich Morbus Crohn und Colitis ulzerosa
Es wurden Fälle einer Neuerkrankung oder einer Exazerbation von entzündlichen Darmerkrankungen mit Taltz berichtet (siehe «Unerwünschte Wirkungen»). Taltz wird für Patienten mit einer entzündlichen Darmerkrankung nicht empfohlen. Wenn ein Patient Anzeichen und Symptome einer entzündlichen Darmerkrankung oder eine Exazerbation einer bereits existierenden entzündlichen Darmerkrankung entwickelt, soll Taltz abgesetzt und eine angemessene medizinische Behandlung eingeleitet werden
Impfungen
Taltz darf nicht zusammen mit Lebendimpfstoff verwendet werden, da keine Daten zu Impfungen mit Lebendimpfstoffen unter Taltz verfügbar sind.
Es wird empfohlen, geplante Impfungen vor Beginn der Therapie mit Taltz abzuschliessen. Dies ist insbesondere bei der Anwendung in der pädiatrischen Population zu beachten. Der zeitliche Abstand zwischen Impfungen mit Lebendimpfstoffen und dem Beginn der Therapie gemäss den aktuellen Impfrichtlinien zu immunsuppressiven Wirkstoffen ist einzuhalten.
In einer offenen Studie an 83 gesunden Probanden mit 1:1 Randomisierung nach einer Kontrollgruppe oder Ixekizumab 160 mg in Woche 0 und 80 mg in Woche 2 wurde die Immunantwort auf zwei inaktivierte Impfstoffe gegen Tetanus (Boostrix®) bzw. Pneumokokken (Pneumovax-23®) untersucht, die in Woche 2 verabreicht wurden. Bis Woche 6 nach Immunisierung wurden in diesem kleinen Kollektiv gesunder Probanden zwar grundsätzlich keine Sicherheitsbedenken festgestellt. Die Extrapolation der Wirksamkeit und Sicherheit inaktivierter Impfungen auf die Zielpopulation ist jedoch nur beschränkt möglich. Die Daten zur Immunantwort waren nicht ausreichend, um auf eine adäquate Immunantwort auf diese Impfstoffe nach der Verabreichung von Taltz in der Zielpopulation schliessen zu können.
Die Impfantwort bei Kindern und Jugendlichen wurde nicht untersucht.
Kombination mit anderen Biologika
Die gleichzeitige Verabreichung von Taltz mit anderen Biologika wurde nicht untersucht und wird nicht empfohlen.
Weitere Hinweise
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro 80 mg Dosis, d.h. es ist nahezu «natriumfrei».
Interaktionen
In Plaque-Psoriasis Studien wurde die Sicherheit von Taltz in Kombination mit anderen Immunmodulatoren oder Phototherapie sowie Impfungen mit Lebendimpfstoffen nicht untersucht.
Cytochrom P450 Substrate
Ergebnisse einer Arzneimittelinteraktionsstudie bei Patienten mit mittelschwerer bis schwerer Psoriasis zeigten, dass die Verabreichung von Ixekizumab mit Arzneimitteln, die durch CYP3A4 (untersucht: Midazolam), CYP2C9 (untersucht: Warfarin), CYP2C19 (untersucht: Omeprazol), CYP1A2 (untersucht: Koffein) oder CYP2D6 (untersucht: Dextromethorphan) metabolisiert werden, keinen klinisch signifikanten Einfluss auf die Pharmakokinetik dieser Arzneimittel hat.
Bei der Verabreichung von Taltz in Kombination mit Methotrexat (MTX), und/oder Kortikosteroiden bei Patienten mit Psoriasis-Arthritis hatten diese Medikamente keinen Einfluss auf die Pharmakokinetik von Taltz.
Die Clearance von Ixekizumab wurde durch die gleichzeitige Gabe von oralen Kortikosteroiden, NSAIDs oder konventionellen DMARDs (z.B. Sulfasalazin und Methotrexat) bei Patienten mit ankylosierender Spondylitis nicht beeinflusst.
Schwangerschaft/Stillzeit
Schwangerschaft
Es gibt begrenzte Daten zur Anwendung von Ixekizumab bei schwangeren Frauen. Es ist bekannt, dass humane IgGs die Plazentaschranke überwinden, und Ixekizumab ist ein IgG. Daher besteht die Möglichkeit, dass Ixekizumab von der Mutter auf den Fötus übergeht. Während der Schwangerschaft und bei gebärfähigen Frauen, die keine wirksame Empfängnisverhütung verwenden, darf Taltz nicht verabreicht werden, es sei denn, dies ist eindeutig erforderlich. Patientinnen sollen angewiesen werden für mindestens 10 Wochen nach der letzten Gabe von Taltz wirksame Verhütungsmethoden anzuwenden.
Tierexperimentelle Studien lassen nicht eindeutig auf direkte oder indirekte schädigende Wirkungen auf Schwangerschaft, embryonale/fetale Entwicklung, Geburt oder postnatale Entwicklung schliessen (siehe «Präklinische Daten»). Da sich anhand tierexperimenteller Reproduktionsstudien nicht immer die Reaktion beim Menschen vorhersagen lässt, sollte Taltz nur dann während einer Schwangerschaft angewendet werden, wenn der Nutzen gegenüber den möglichen Risiken eindeutig überwiegt.
Stillzeit
Es ist nicht bekannt, ob Ixekizumab beim Menschen in die Muttermilch übertritt oder nach oraler Aufnahme systemisch resorbiert wird. Allerdings geht Ixekizumab bei Cynomolgus Affen in geringen Mengen in die Muttermilch über. Da viele Medikamente, darunter Antikörper, in die Muttermilch ausgeschieden werden, kann ein Risiko für das Neugeborene/Kleinkind nicht ausgeschlossen werden. Wegen möglichen Schaden für den gestillten Säugling wird es empfohlen während der Behandlung mit Taltz und während mindestens 10 Wochen nach der letzten Dosis nicht zu stillen. Eine Entscheidung, entweder mit dem Stillen aufzuhören oder die Behandlung mit Taltz abzubrechen sollte unter Berücksichtigung der Vorteile des Stillens für das Kind und der Vorteile der Therapie für die Mutter, gefällt werden.
Fertilität
Die Wirkung von Ixekizumab auf die humane Fertilität wurde nicht untersucht. Tierstudien weisen nicht auf direkte oder indirekte schädliche Wirkungen auf die Fertilität hin (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Der Einfluss von Taltz auf die Fahrtüchtigkeit oder die Fähigkeit zum Bedienen von Maschinen wurde nicht untersucht.
Unerwünschte Wirkungen
Insgesamt wurden 8956 Patienten in verblindeten und offenen klinischen Studien zu Plaque-Psoriasis, Psoriasis-Arthritis, ankylosierender Spondylitis und anderen Indikationen mit Taltz behandelt. Hiervon hatten 6385 Patienten Taltz über mindestens ein Jahr hinweg erhalten, was kumulativ einer Exposition von 19'833 erwachsenen Patientenjahren und 207 pädiatrischen Patientenjahren (196 pädiatrische Patienten) entspricht.
Erwachsene
Bei Plaque-Psoriasis in Erwachsenen wurden drei placebokontrollierte Studien Phase III integriert ausgewertet, um die Sicherheit von Taltz über 12 Wochen nach Therapiebeginn im Vergleich zu Placebo zu untersuchen. Insgesamt 3119 Patienten wurden ausgewertet (1161 Patienten unter 80 mg alle 4 Wochen (Q4W), 1167 Patienten unter 80 mg alle 2 Wochen (Q2W) und 791 Patienten unter Placebo).
In zwei dieser Phase III Studien wurde die Sicherheit von Taltz auch mit Etanercept über 12 Wochen ab Therapiebeginn (729 Patienten unter 80 mg Q4W, 734 Patienten unter 80 mg Q2W und 739 Patienten unter Etanercept) verglichen.
Das beobachtete Sicherheitsprofil bei pädiatrischen Patienten mit Plaque-Psoriasis, die alle 4 Wochen mit Taltz behandelt werden, stimmt insgesamt mit demjenigen bei Erwachsenen überein (siehe separaten Abschnitt zur Pädiatrie).
Bei Psoriasis-Arthritis in Erwachsenen wurden zwei placebokontrollierte Studien Phase III integriert ausgewertet, um die Sicherheit von Taltz über 24 Wochen nach Therapiebeginn im Vergleich zu Placebo zu untersuchen. Insgesamt 678 Patienten wurden ausgewertet (229 Patienten unter 80 mg alle 4 Wochen (Q4W), 225 Patienten unter 80 mg alle 2 Wochen (Q2W) und 224 Patienten unter Placebo). Das Sicherheitsprofil für Patienten mit Psoriasis-Arthritis ist vergleichbar mit dem für Plaque-Psoriasis-Patienten.
Bei der ankylosierenden Spondylitis wurde Taltz in zwei placebokontrollierten Studien untersucht. Insgesamt wurden 657 Patienten untersucht (376 Patienten mit Taltz und 191 mit Placebo). Insgesamt erhielten 195 Patienten in diesen Studien Taltz 80 oder 160 mg in Woche 0, gefolgt von 80 mg alle 4 Wochen (Q4W). Insgesamt ist das beobachtete Sicherheitsprofil bei Patienten mit ankylosierender Spondylitis mit Taltz Q4W vergleichbar mit Plaque-Psoriasis-Patienten.
Die häufigsten berichteten unerwünschten Wirkungen waren Reaktionen an der Injektionsstelle und Infektionen der oberen Atemwege (einschliesslich Nasopharyngitis und Pharyngitis). Die meisten Reaktionen waren von leichtem oder mittlerem Schweregrad.
Liste der Nebenwirkungen in placebo-kontrollierten klinischen Studien (Phase III, integrierte Daten aus des Indikationen Plaque Psoriasis, Psoriasis-Arthritis, und ankylosierende Spondylitis) und Meldungen nach Markteinführung
Die Häufigkeiten der unerwünschten Wirkungen werden wie folgt angegeben: «sehr häufig» (≥1/10), «häufig» (≥1/100, <1/10), «gelegentlich» (≥1/1000, <1/100), «selten» (≥1/10'000, <1/1000) und «sehr selten» (<1/10'000).
Infektionen und Infestationen
Sehr häufig: Infektionen der oberen Atemwege (13.4% mit Q2W und 13.5% mit Q4W).
Häufig: Tinea Infektion.
Gelegentlich: Influenza, Rhinitis, orale Candidiasis, Konjunktivitis.
Selten: oesophagale Candidiasis.
Störungen des Blut- und Lymphsystems
Gelegentlich: Neutropenie, Thrombozytopenie.
Störungen des Immunsystems
Selten: Anaphylaxie (Angioödem, Larynxödem, Urtikaria).
Respiratorische, thorakale und mediastinale Funktionsstörungen
Häufig: oropharyngeale Schmerzen.
Gastrointestinale Störungen
Häufig: Übelkeit, Diarrhö.
Gelegentlich: Morbus Crohn, Colitis ulzerosa (siehe «Warnhinweise und Vorsichtsmassnahmen»)
Leber- und Gallenerkrankungen
Häufig: erhöhte Leberenzyme.
Funktionsstörungen der Haut und des Unterhautzellgewebes
Gelegentlich: Urtikaria.
Allgemeine Störungen und Reaktionen an der Applikationsstelle
Sehr häufig: Reaktionen an der Injektionsstelle (18.2% mit Q2W und 13.7% mit Q4W).
Beschreibung ausgewählter Nebenwirkungen
Reaktionen an der Injektionsstelle
Die am häufigsten an der Injektionsstelle beobachteten Reaktionen waren Erythem und Schmerzen. Diese Reaktionen waren vorwiegend von leichtem bis mittlerem Schweregrad und führten nicht zum Abbruch von Taltz.
Infektionen
Die Mehrzahl der Infektionen bestand aus nicht-schwerwiegenden und leichten bis mittelschweren unerwünschten Ereignissen wie Nasopharyngitis und Infektionen der oberen Atemwege, die einen Therapieabbruch nicht erforderlich machten.
In den placebokontrollierten Phasen III klinischen Studien bei Plaque Psoriasis in Erwachsenen (insgesamt wurden 2328 Patienten mit Taltz und 791 Patienten mit Placebo über bis zu 12 Wochen behandelt) wurden Infektionen bei 27.2 % der Patienten unter Taltz im Vergleich zu 22.9 % der Patienten unter Placebo berichtet.
Schwerwiegende Infektionen traten bei 13 (0.6 %) der Patienten unter Taltz und bei 3 (0.4 %) der Patienten unter Placebo auf (siehe «Warnhinweise und Vorsichtsmassnahmen»). Mit Infektionen zusammenhängende schwerwiegende unerwünschte Ereignisse (SAE), die von mehr als 1 Patient der gesamten Ixekizumab-Gruppe berichtet wurden, waren Cellulitis (n = 3), Appendizitis (n = 2) und Erysipel (n = 2). Der Anteil der Patienten, die die Therapie aufgrund eines mit Infektionen zusammenhängenden unerwünschten Ereignisses abbrach, war in der gesamten Ixekizumab-Gruppe (8 Patienten [0.3 %]) und in der Placebo-Gruppe (2 Patienten [0.3 %]) ähnlich.
Übereinstimmend mit dem Wirkmechanismus zeigte sich ein Anstieg oraler Candidiasis und mit einer Ausnahme waren alle Fälle von leichtem oder mittlerem Schweregrad. Es wurde über keine SAE oder Therapieabbrüche aufgrund von Candidiasis berichtet.
Über die gesamte Behandlungsdauer wurden Infektionen bei 52.8 % der Patienten unter Taltz berichtet (46.9 pro 100 Patientenjahre). Schwerwiegende Infektionen wurden von 1.6 % der Patienten unter Taltz berichtet (1.5 pro 100 Patientenjahre).
Neutropenie und Thrombozytopenie
In Plaque-Psoriasis Studien entwickelten 9 % der mit Taltz behandelten Patienten eine Neutropenie. Ein solcher Grad der Neutropenie kann weiterbestehen, fluktuieren oder vorübergehend sein.
0.1 % der mit Taltz behandelten Patienten entwickelten eine Neutrophilenzahl von < 1000 Zellen/mm³. Im Allgemeinen erforderte die Neutropenie kein Absetzen von Taltz.
3 % der mit Taltz behandelten Patienten mit einem normalen Ausgangswert der Thrombozytenzahl zeigten eine Verminderung dieser Zahl auf Werte zwischen 75'000 und 150'000 Zellen/mm³. Die Thrombozytopenie kann weiterbestehen, fluktuieren oder vorübergehend sein.
Die Häufigkeit von Neutropenie und Thrombozytopenie in den klinischen Studien zur Psoriasis-Arthritis ist mit der bei Plaque-Psoriasis-Studien beobachteten Häufigkeit vergleichbar.
Sicherheit im Vergleich zu Etanercept
In den 2 aktiv kontrollierten klinischen Studien UNCOVER-2 und UNCOVER-3 waren die Raten für schwerwiegende unerwünschte Ereignisse sowohl für Taltz als auch für Etanercept 1.9% und die Abbruchraten aufgrund von unerwünschten Ereignissen waren 1.2% für Etanercept und 2% für Taltz. Die Infektionsrate war 21.5% für Etanercept und 26% für Taltz wobei der Schweregrad für die meisten Ereignisse mild bis mässig war. Die Rate für schwere Infektionen lag bei Etanercept bei 0.4% und bei Taltz bei 0.5%.
Pädiatrie
Insgesamt stimmt das Sicherheitsprofil, das bei pädiatrischen Patienten mit Plaque-Psoriasis, die alle 4 Wochen mit Taltz behandelt werdenmit dem Sicherheitsprofil bei erwachsenen Patienten mit Plaque-Psoriasis überein, mit Ausnahme folgender unerwünschter Wirkungen, die im Vergleich zu Erwachsenen häufiger beobachtet wurden, oder aber spezifisch bei Kindern und Jugendlichen auftraten:
Infektionen und Infestationen
Häufig: Influenza, Konjunktivitis.
Störungen des Blut- und Lymphsystems
Häufig: Neutropenie.
Störungen des Immunsystems
Häufig: Überempfindlichkeitsreaktionen (Bronchospasmus, Dermatitis, Rash).
Funktionsstörungen der Haut und des Unterhautzellgewebes
Häufig: Urtikaria.
Allgemeine Störungen und Reaktionen an der Applikationsstelle
Häufig: Fieber.
Beschreibung ausgewählter Nebenwirkungen
Neutropenie
In der pädiatrischen Studie in 114 Patienten im Alter von 6-17 mit mittelschwerer oder schwerer Plaque-Psoriasis entwickelten 8.8 % der mit Taltz behandelten Patienten eine Neutropenie, welche weiterbestehen, fluktuieren oder transient auftreten können.
0.9 % der mit Taltz behandelten Patienten entwickelten eine Neutrophilenzahl von < 1000 Zellen/mm³. In der pädiatrischen Studie wurde Taltz bei keinem Patienten wegen Neutropenie abgesetzt.
Überempfindlichkeitsreaktionen
In der pädiatrischen Studie traten während des 12-wöchigen, placebokontrollierten Zeitraums bei 5.2% der Patienten in der Taltz-Gruppe und 1.8% der Patienten in der Placebo-Gruppe, Überempfindlichkeitsreaktionen auf. Alle Überempfindlichkeitsreaktionen waren nichtanaphylaktischer Natur.
Entzündliche Darmerkrankungen.
Entzündliche Darmerkrankungen wurden bei pädiatrischen Patienten gelegentlich beobachtet und waren jedoch häufiger als bei Erwachsenen.
In der pädiatrischen Studie trat während des 12-wöchigen, placebokontrollierten Zeitraums ein Morbus Crohn bei 0.9% der Patienten in der Taltz-Gruppe und bei 0% der Patienten in der Placebo-Gruppe auf. Über die Gesamtlaufzeit der pädiatrischen Studie (Placebo-kontrollierte Phase und Erhaltungstherapie Phase) trat ein Morbus Crohn bei insgesamt 4 mit Taltz behandelten Probanden (2.0%) auf.
Reaktionen an der Injektionsstelle
In der pädiatrischen Plaque-Psoriasis Studie traten, während des 12-wöchigen placebokontrollierten Zeitraums, bei 12.2% der Patienten in der Taltz-Gruppe und 1.8% der Patienten in der Placebo-Gruppe, Reaktionen an der injektionsstelle auf. Die am häufigsten an der Injektionsstelle beobachteten Reaktionen waren Schmerzen und Erythem. Diese Reaktionen waren vorwiegend von leichtem bis mittlerem Schweregrad und führten nicht zum Abbruch von Taltz.
Immunogenität
Etwa 9 – 17 % der mit der empfohlenen Taltz Dosis behandelten Plaque-Psoriasis erwachsenen Patienten entwickelten Antikörper gegen Ixekizumab, bei der Mehrzahl waren die Titer niedrig und in der bis zu 60 Wochen dauernden Therapie nicht mit einem verminderten klinischen Ansprechen verbunden. Etwa 1 % der mit Taltz behandelten Patienten hatten jedoch bestätigte, neutralisierende Antikörper, die mit niedrigen Arzneimittel-Konzentrationen und reduziertem klinischen Ansprechen verbunden waren.
Von den pädiatrischen Patienten mit Plaque-Psoriasis, die mit Taltz im empfohlenen Dosierschema bis zu 12 Wochen behandelt wurden, entwickelten 21 Patienten (18%) Antikörper gegen den Wirkstoff.
Bei ungefähr der Hälfte waren die Titer niedrig, und 5 Patienten (4%) hatten bestätigte neutralisierende Antikörper, die mit niedrigen Arzneimittel-Konzentrationen verbunden waren. Es gab keinen Zusammenhang mit klinischem Ansprechen oder unerwünschten Ereignissen.
Von den Psoriasis-Arthritis Patienten, die mit Taltz im empfohlenen Dosierschema bis zu 52 Wochen behandelt wurden, entwickelten 11% Antikörper gegen den Wirkstoff, wovon die Mehrheit einen niedrigen Titer aufwies, und 8% hatten nachweislich neutralisierende Antikörper. Die Häufigkeit von Antikörpern gegen den Wirkstoff und neutralisierenden Antikörpern betrug 15.4% bzw. 10.3% in der Gruppe ohne gleichzeitige Gabe von cDMARDs gegenüber 9.0% bzw. 6.0% in der Gruppe, die Ixekizumab in Kombination mit cDMARDs erhielt. Es konnte kein eindeutiger Zusammenhang zwischen der Anwesenheit von neutralisierenden Antikörpern und einem Einfluss auf die Wirkstoffkonzentration oder die Wirksamkeit beobachtet werden.
Bei Patienten mit ankylosierender Spondylitis, die mit Taltz im empfohlenen Dosierschema behandelt wurden, entwickelten bis Woche 16 und Woche 52 10 Patienten (5.2%) bzw. 23 Patienten (11.9 %) Antikörper gegen den Wirkstoff, wovon die Mehrheit einen niedrigen Titer aufwies; 3 Patienten (1.5%) hatten neutralisierende Antikörper bis Woche 16, und keine neuen Patienten entwickelten neutralisierende Antikörper zwischen Woche 16 und Woche 52. Bei einigen Antikörper gegen den Wirkstoff-positiven Patienten wurden tendenziell niedrigere Talspiegel beobachtet. Es konnte kein offensichtlicher Zusammenhang zwischen der Anwesenheit von Antikörpern gegen den Wirkstoff und einem Einfluss auf die Wirksamkeit oder die Sicherheit beobachtet werden.
Bei allen Indikationen, wurde ein Zusammenhang zwischen Immunogenität und während der Therapie aufgetretenen unerwünschten Ereignissen nicht festgestellt.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
In klinischen Studien wurden Dosierungen bis zu 180 mg subkutan ohne Auftreten dosislimitierender Toxizitäten angewendet. Es wurde über Überdosierungen bis zu 240 mg subkutan berichtet, ohne dass schwerwiegende unerwünschte Ereignisse auftraten Bei Überdosierung wird empfohlen, Patienten auf Anzeichen und Symptome unerwünschter Wirkungen zu überwachen und unverzüglich eine angemessene symptomatische Behandlung einzuleiten.
Eigenschaften/Wirkungen
ATC-Code
L04AC13
Wirkungsmechanismus
Ixekizumab ist ein für Interleukin-17A selektiver, rekombinanter, humanisierter, monoklonaler Antikörper. Ixekizumab ist ein aus CHO (Chinese Hamster Ovary) - Zellen hergestellter modifizierter monoklonaler IgG4 Antikörper, der mit hoher Affinität (< 3 pM) und Spezifität an das proinflammatorische Zytokin Interleukin 17A (sowohl an das IL-17A als auch das Heterodimer IL-17A/F) bindet. Erhöhte Werte von IL-17A wurden mit der Pathogenese verschiedener Autoimmunerkrankungen in Verbindung gebracht. Bei Psoriasis spielt der IL-17A-Ligand eine wichtige Rolle bei der überschiessenden Keratinozytenproliferation und -aktivierung. Die Neutralisierung von IL-17A durch Ixekizumab hemmt diese Wirkungen. IL-17A spielt eine Schlüsselrolle in der Pathogenese von Plaque-Psoriasis und Psoriasis-Arthritis und ist hochreguliert in von Läsionen betroffener Haut im Gegensatz zu nicht von Läsionen betroffener Haut von Plaque Psoriasis Patienten.
Bei der ankylosierenden Spondylitis spielt der IL-17A-Ligand eine wichtige Rolle durch die Zunahme der Entzündungen, die zu erosiven Knochenschäden und pathologischer Knochenneubildung führt.
Ixekizumab bindet nicht an die Liganden IL-17B, IL-17C, IL-17D, IL-17E oder IL-17F.
Ixekizumab hat eine geringe Fähigkeit zu Bindung an Fcγ Rezeptoren oder Komponenten des Komplementsystems. In-vitro durchgeführte Bindungsversuche bestätigten, dass Ixekizumab nicht an die humanen Fcγ Rezeptoren I, IIa und IIIa oder die Komplementkomponente C1q bindet.
Pharmakodynamik
Auf Grundlage der Daten einer Phase I Studie mit Biopsien, die aus Psoriasis-betroffener Haut entnommen wurden, gab es vom Ausgangswert bis Tag 43 einen dosisabhängigen Trend zur Verringerung von Epidermisdicke, Anzahl proliferierender Keratinozyten, T-Zellen und dendritischer Zellen sowie Verringerung der lokalen Entzündungsmarker. Als direkte Folge reduziert die Behandlung mit Ixekizumab Erythem, Verhärtung und Schuppung in Plaque-Psoriasis-Läsionen.
Taltz hat eine Senkung des C-reaktiven Proteins-Levels (innerhalb von 1 Woche Behandlung) gezeigt.
Klinische Wirksamkeit
Plaque-Psoriasis
Die Wirksamkeit und Sicherheit von Taltz wurde in drei randomisierten, doppelblinden, placebokontrollierten Phase III Studien bei erwachsenen Patienten mit mittelschwerer bis schwerer Plaque Psoriasis untersucht, die Kandidaten für eine Phototherapie oder systemische Therapie waren (UNCOVER-1, UNCOVER-2 und UNCOVER-3). Die Wirksamkeit und Sicherheit von Taltz wurde auch im Vergleich zu Etanercept untersucht (UNCOVER-2 und UNCOVER-3). Patienten, die ursprünglich auf Taltz randomisiert wurden und die in Woche 12 einen static Physician Global Assesment Responder -Wert von 0 oder 1 (sPGA (0,1)) erreichten, wurden erneut auf Placebo oder Taltz für weitere 48 Wochen (UNCOVER-1 und UNCOVER-2) randomisiert. Auf Placebo, Etanercept oder Taltz randomisierte Patienten die sPGA (0,1) non Responder waren, erhielten Taltz für bis zu 48 Wochen.
Von den 3866 in diese placebokontrollierten Studien eingeschlossenen Patienten hatten 64 % zuvor eine systemische Therapie erhalten (biologische, konventionelle systemische oder PUVA), 43.5 % hatten zuvor eine Phototherapie erhalten, 49.3 % eine konventionelle systemische Therapie und 26.4 % eine biologische Therapie der Psoriasis. Von allen Patienten hatten 14.9 % mindestens eine anti-TNF-alpha Behandlung bekommen, und 8.7 % eine anti-IL-12/IL-23 Behandlung. Zu Studienbeginn hatten 23.4 % der Patienten eine Psoriasis-Arthritis in der Vorgeschichte.
Ausschlusskriterien: wesentliche Ausschlusskriterien, die einer Teilnahme an UNCOVER-1, UNCOVER-2 und UNCOVER-3 entgegenstanden, waren die begleitende Anwendung von systemischen oder biologischen Psoriasis-Therapien oder Phototherapie, jede instabile schwerwiegende medizinische Störung oder Erkrankung ausgenommen Plaque Psoriasis, und jede derzeit bestehende oder kürzlich bestandene schwerwiegende Infektion. In UNCOVER-2 und UNCOVER-3 war jede vorherige Anwendung von Etanercept ein Ausschlusskriterium.
Die kombinierten primären Endpunkte in allen drei Studien waren der Anteil Patienten im Vergleich zu Placebo, die in Woche 12 ein PASI-75-Ansprechen (Psoriasis Area and Severity Index; PASI 75 = Verbesserung des PASI um mindestens 75 %) und einen sPGA-Wert von 0 («frei von») oder 1 («minimal») erreichten. Der Baseline Medianwert des PASI für Patienten aller Therapiegruppen lag zwischen 17.4 und 18.3; 48.3 % bis 51.2 % der Patienten hatten einen Baseline sPGA-Wert von «schwer» oder «sehr schwer», und der Medianwert der itch Numeric Rating Scale (itch NRS) lag bei Baseline zwischen 6.3 und 7.1.
Tabelle 1. Klinisches Ansprechen nach 12 Wochen (NRI) aus UNCOVER-1, -2 und -3
Placebo | Taltz 80 mg Q4W | Taltz | Etanercept | |
|---|---|---|---|---|
UNCOVER 1 | ||||
Anzahl Patienten (N) | 431 | 432 | 433 | NA |
sPGA «0» (frei von) oder «1» (minimal), n (%) | 14 (3.2%) | 330 (76.4%)a | 354 (81.8%)a | NA |
sPGA «0» (frei von), n (%) | 0 | 149 (34.5%)a | 160 (37.0%)a | NA |
PASI 75, n (%) | 17 (3.9%) | 357 (82.6%)a | 386 (89.1%)a | NA |
PASI 90, n (%) | 2 (0.5%) | 279 (64.6%)a | 307 (70.9%)a | NA |
PASI 100, n (%) | 0 | 145 (33.6%)a | 153 (35.3%)a | NA |
UNCOVER 2 | ||||
Anzahl Patienten (N) | 168 | 347 | 351 | 358 |
sPGA «0» (frei von) oder «1» (minimal), n (%) | 4 (2.4%) | 253 (72.9%)a | 292 (83.2%)a | 129 (36.0%)a |
sPGA «0» (frei von), n (%) | 1 (0.6%) | 112 (32.3%)a,b | 147 (41.9%)a,b | 21 (5.9%)c |
PASI 75, n (%) | 4 (2.4%) | 269 (77.5%)a | 315 (89.7%)a | 149 (41.6%)a |
PASI 90, n (%) | 1 (0.6%) | 207 (59.7%)a,b | 248 (70.7%)a,b | 67 (18.7%)a |
PASI 100, n (%) | 1 (0.6%) | 107 (30.8%)a,b | 142 (40.5%)a,b | 19 (5.3%)c |
UNCOVER 3 | ||||
Anzahl Patienten (N) | 193 | 386 | 385 | 382 |
sPGA «0» (frei von) oder «1» (minimal), n (%) | 13 (6.7%) | 291 (75.4%)a,b | 310 (80.5%)a,b | 159 (41.6%)a |
sPGA «0» (frei von), n (%) | 0 | 139 (36.0%)a,b | 155 (40.3%)a,b | 33 (8.6%)a |
PASI 75, n (%) | 14 (7.3%) | 325 (84.2%)a,b | 336 (87.3%)a,b | 204 (53.4%)a |
PASI 90, n (%) | 6 (3.1%) | 252 (65.3%)a,b | 262 (68.1%)a,b | 98 (25.7%)a |
PASI 100, n (%) | 0 | 135 (35.0%)a,b | 145 (37.7%)a,b | 28 (7.3%)a |
Abkürzungen: n = Anzahl Patienten in dieser Kategorie; N = Anzahl Patienten in der Intent-to-treat Population; NA = nicht zutreffend; NRI = Non-Responder Imputation; NRS = numeric rating scale; PASI = Psoriasis Area and Severity Index; Q2W = alle zwei Wochen; Q4W = all vier Wochen; sPGA = static Physician Global Assessment.
a p < 0.001 im Vergleich zu Placebo
b p < 0.001 im Vergleich zu Etanercept
c p < 0.01 im Vergleich zu Placebo
UNCOVER-1 schloss 1296 Patienten ein. Die Patienten wurden randomisiert (1:1:1) auf 12 Wochen Placebo oder Taltz (80 mg alle zwei oder vier Wochen [Q2W oder Q4W] nach einer Startdosis von 160 mg).
UNCOVER-2 schloss 1224 Patienten ein. Die Patienten wurden randomisiert (1:2:2:2) auf entweder 12 Wochen Placebo oder Taltz (80 mg alle zwei oder vier Wochen [Q2W oder Q4W] nach einer Startdosis von 160 mg) oder Etanercept 50 mg 2x wöchentlich.
UNCOVER-3 schloss 1346 Patienten ein. Die Patienten wurden randomisiert (1:2:2:2) auf entweder 12 Wochen Placebo oder Taltz (80 mg alle zwei oder vier Wochen [Q2W oder Q4W] nach einer Startdosis von 160 mg) oder Etanercept 50 mg 2x wöchentlich.
Die Dosierung 80 mg Q2W zeigte in allen Studien bei allen Endpunkten eine überlegene Wirksamkeit (siehe Tabellen oben), insbesondere bei hohen Graden der Hautverbesserung (PASI 90, PASI 100, sPGA 0) und bei der Reduktion des Juckreizes. Im Vergleich zu Placebo und Etanercept wurden in Woche 12 signifikant grössere Verbesserungen des Ausgangswerts gezeigt, bei Nagel Psoriasis (gemessen anhand des Nail Psoriasis Severity Index (NAPSI)), bei Kopfhaut Psoriasis (gemessen anhand des Psoriasis Scalp Severity Index (PSSI)) und bei palmoplantarer Psoriasis (gemessen anhand des Psoriasis Palmoplantar Severity Index (PPASI)).
Taltz war mit einem raschen Einsetzen der Wirkung mit einer Reduktion des mittleren PASI in Woche 2 um > 50% verbunden (Abbildung 1). Der Anteil der Patienten, der PASI 75 erreichte, war bereits in Woche 1 in allen drei Studien unter Taltz signifikant grösser als unter Placebo und unter Etanercept. Etwa 25 % der mit Taltz behandelten Patienten erreichte bis Woche 2 einen PASI-Wert< 5, mehr als 55 % erreichten einen PASI-Wert < 5 bis Woche 4 mit einem Anstieg auf 85 % bis Woche 12 (im Vergleich zu 3 %, 14 % und 50 % unter Etanercept). Signifikante Verbesserungen des Juckreiz-Schweregrades wurden in Woche 1 unter Taltz beobachtet.
Taltz zeigte auch einen günstigen Einfluss auf den Pruritus, der Placebo signifikant überlegen war.
Abbildung 1. PASI Werte, prozentuale Verbesserung bei jeder Visite nach der Baseline Visite (mBOCF) in der Intent-to-treat Population während der Anfangsphase der Therapie (UNCOVER-2 und UNCOVER-3)

Die Wirksamkeit und Sicherheit von Taltz wurde unabhängig von Alter, Geschlecht, ethnischer Abstammung, Körpergewicht, PASI Baseline Schweregrad und vorangegangener Therapie mit einem Biologikum, gezeigt. Die Ansprechrate nach 12 Wochen für mit Taltz 80 mg Q2W behandelten Patienten < 100 kg bzw. ≥100kg waren PASI 75 (90.1% bzw. 85.7%) und sPGA 0 oder 1 (84.5% bzw. 75.6%). Die Ansprechrate nach 12 Wochen für mit Taltz 80 mg Q4W behandelten Patienten < 100 kg bzw. ≥100kg waren PASI 75 (85.6% bzw. 74.2%) und sPGA 0 oder 1 (79.0% bzw. 67.4%). Das Ansprechen unterschied sich nicht bei Patienten mit Nagel Psoriasis, Psoriasis im Gesichtsbereich oder Kopfhaut Psoriasis zu Studienbeginn. Taltz war wirksam bei Patientenmit früheren systemischen Therapien, mit oder ohne vorherige Biologika/Anti-TNF-Exposition einschliesslich Patienten, die ein Therapieversagen unter einer Biologika/anti-TNF-Behandlung aufwiesen. Die Verbesserungen von sPGA und PASI Endpunkten bei Patienten mit begleitender Psoriasis-Arthritis zu Studienbeginn waren ähnlich zu jenen der gesamten Population mit mittelschwerer bis schwerer Plaque Psoriasis. Etwa 45 % der Patienten wiesen bei Studienbeginn Psoriasis im Gesichtsbereich auf. Von diesen Patienten waren 80.4 % der mit Taltz behandelten Patienten in Woche 12 frei von Psoriasis im Gesichtsbereich.
Wirksamkeit bei Etanercept-Non-Respondern: Bei Patienten, die in Woche 12 der UNCOVER-2 als Etanercept-sPGA (0,1) Non-Responder angesehen wurden (N = 200) und die nach einer 4-wöchigen Auswaschphase auf Taltz 80 mg Q4W umgestellt wurden, erreichten nach 12 Wochen 73 % einen sPGA (0,1) und 83.5% einen PASI 75.
Anhalten des Ansprechens bis Woche 60
Zur Beurteilung des anhaltenden Ansprechens wurden Patienten aus UNCOVER-1 und UNCOVER-2, die ursprünglich auf Taltz randomisiert worden waren und in Woche 12 angesprochen hatten (d.h. sPGA-Werte von 0 oder 1) erneut auf eine der folgenden Therapien für weitere 48 Wochen, randomisiert: Placebo oder Taltz (80 mg alle vier oder zwölf Wochen [Q4W oder Q12W]). Patienten, die in Woche 12 einen sPGA (0,1) nicht erreicht hatten oder während der Erhaltungsphase einen Rückfall (sPGA ≥3) erlitten, erhielten anschliessend Taltz 80 mg Q4W.
Bei den sPGA (0,1) Respondern in Woche 12 (der kombinierten Studien UNCOVER-1 und UNCOVER-2) war der Anteil der Patienten, der dieses Ansprechen in Woche 60 aufrecht erhalten hatte bei Patienten unter Taltz 80 mg Q4W (71 %) signifikant grösser im Vergleich zu Patienten unter Taltz 80 mg Q12W (35.5 %) oder Placebo (7 %).
Tabelle 2 zeigt die Ansprechraten der bei der erneuten Randomisierung auf die empfohlene Erhaltungsdosis von 80 mg Taltz alle 4 Wochen randomisierten Patienten in Abhängigkeit der randomisierten Dosis zu Studienbeginn.
Tabelle 2. Anhaltendes Ansprechen und Wirksamkeit in Woche 60 (kombinierte Ergebnisse aus den Studien UNCOVER-1 und UNCOVER-2; NRI)
Endpunkt | 80 mg Q2W (Induktion)/Placebo (Erhaltung) (N=211) | 80 mg Q4W (Induktion)/ Placebo (Erhaltung) (N=191) | 80 mg Q2W (Induktion)/80 mg Q4W (Erhaltung) (N=221) | 80 mg Q4W (Induktion)/ 80 mg Q4W (Erhaltung) (N=195) |
|---|---|---|---|---|
Beibehalten eines sPGA «0» (frei von) oder «1» (minimal) | 7.6 % | 6.3 % | 78.3 % | 68.7 % |
sPGA «0» (frei von) beibehalten oder erreicht | 2.8 % | 1.6 % | 58.8 % | 49.2 % |
PASI 75 | 9.0 % | 7.9 % | 83.3 % | 74.4 % |
PASI 90 | 4.7 % | 4.7 % | 76.5 % | 66.7 % |
PASI 100 | 2.8 % | 1.6 % | 57.5 % | 49.7 % |
Abkürzungen: N = Anzahl Patienten in der Analysenpopulation; NRI = Non-Responder Imputation
Von den sPGA (0,1) Respondern in Woche 12, die der Erhaltungstherapie Taltz 80 mg Q4W zugeordnet wurden, hatten, in Woche 60, 76.4 % einen PASI < 5 beibehalten oder erreicht. Die Verbesserung des Juckreiz-Schweregrades hielt bei den Taltz Patienten, die in Woche 12 zu den sPGA (0,1) Respondern gehörten, bis Woche 60 an. Im Hinblick auf das anhaltende Ansprechen bis Woche 60 war Taltz wirksam bei Patienten mit früherer systemischer Therapie, mit oder ohne vorherige Biologika/Anti-TNF-Exposition einschliesslich Patienten, die ein Therapieversagen unter einer Biologika/anti-TNF-Behandlung aufwiesen.
Von den sPGA (0,1) Respondern in Woche 12, die Placebo rerandomisiert wurden, betrug die Zeit bis zum Rückfall (sPGA ≥3) in den kombinierten UNCOVER-1 und UNCOVER-2 Studien 164 (95% CI [143, 169]) Tage. Von diesen Patienten erreichten 71.5 % innerhalb von 12 Wochen nach erneutem Beginn mit Taltz 80 mg Q4W wieder einen sPGA-Wert von 0 oder 1.
Bei Patienten unter Taltz, die in Woche 12 zu den sPGA (0,1) Respondern gehörten, hielten auch die Verbesserungen bei Nagel Psoriasis, Kopfhaut Psoriasis und palmoplantarer Psoriasis bis Woche 60 an.
Quality of life/Patientenberichtete Ergebnisse
Für den DLQI (Dermatology Life Quality Index) wurden statistisch signifikante Verbesserungen in Woche 12 (Studien 1-3) gegenüber dem Ausgangswert gezeigt; die Verbesserungen blieben über 60 Wochen bestehen Taltz war mit signifikant grösseren Verbesserungen der Hautschmerzen (gemessen anhand der visuellen Analogskala, VAS) verbunden.
Die Wirksamkeit und Sicherheit von Ixekizumab wurden auch in der doppelblinden Studie RHBS im Vergleich zu Ustekinumab untersucht. Dabei war Ixekizumab im Hinblick auf den primären Endpunkt (PASI 90-Ansprechen in Woche 12, Tabelle 3) überlegen.). In allen drei Kategorien des PASI-Ansprechens zeigte sich die Überlegenheit versus Ustekinumab schnell. Die Überlegenheit von Ixekizumab versus Ustekinumab wurde ebenfalls in den nach Gewicht stratifizierten Subgruppen gezeigt.
Tabelle 3: PASI-Ansprechraten aus der Vergleichsstudie Ixekizumab versus Ustekinumab
Woche 12 | Woche 52 | |||
|---|---|---|---|---|
Ixekizumab* | Ustekinumab** | Ixekizumab* | Ustekinumab** | |
Patienten (n) | 136 | 166 | 136 | 166 |
PASI 75, n (%) | 120 (88.2 %) | 114 (68.7 %) | 120 (88.2 %) | 126 (75.9 %) |
PASI 90, n (%) | 99 (72.8 %)§ | 70 (42.2 %) | 104 (76.5 %) | 98 (59.0 %) |
PASI 100, n (%) | 49 (36.0 %) | 24 (14.5 %) | 71 (52.2 %) | 59 (35.5 %) |
* Ixekizumab 160 mg wurde als Initialdosis verabreicht, gefolgt von 80 mg in den Wochen 2, 4, 6, 8, 10 und 12, und anschliessend 80 mg Q4W
** Gewichtsbasierte Dosierung: Patienten, die mit Ustekinumab behandelt wurden, erhielten 45 mg oder 90 mg in Woche 0 und 4, anschliessend alle 12 Wochen bis Woche 52 (dosiert nach Gewicht gemäss zugelassener Dosierung)
§ p < 0.001 versus Ustekinumab (p-Wert nur für den primären Endpunkt)
Wirksamkeit bei genitaler Psoriasis
Eine randomisierte, doppelblinde, placebokontrollierte Studie (IXORA-Q) wurde an 149 erwachsenen Patienten (24 % Frauen) mit einer mittelschweren bis schweren genitalen Psoriasis (sPGA des Genitalbereichs von ≥3) durchgeführt. Die Patienten hatten eine Hautbeteiligung von mindestens 1 % Body Surface Area (BSA) (60.4 % hatten eine Hautbeteiligung von ≥10% BSA) und haben auf mindestens eine vorangegangene topische Therapie zur Behandlung der genitalen Psoriasis nicht angesprochen oder diese nicht vertragen. Die Patienten hatten zumindest eine mittelschwere Plaque-Psoriasis (definiert als sPGA-score ≥3 und waren geeignet für eine Phototherapie und/oder systemische Therapie) über mindestens 6 Monate.
Studienteilnehmer, die auf Taltz randomisiert wurden, erhielten eine Initialdosis von 160 mg, gefolgt von 80 mg alle 2 Wochen für eine Dauer von 12 Wochen. Der primäre Endpunkt war der Anteil an Patienten, die einen sPGA im Genitalbereich von «0» (frei von) oder «1» (minimal) (sPGA-G 0/1) erreichten. In Woche 12 erreichten, unabhängig von der Hautbeteiligung zu Studienbeginn, signifikant mehr Studienteilnehmer unter Taltz einen sPGA-G 0/1 und einen sPGA 0/1 als Studienteilnehmer unter Placebo (Hautbeteiligung zu Studienbeginn von 1 % bis < 10 % BSA bzw. ≥10 % BSA: sPGA-G von «0» oder «1»: Taltz 71 % bzw. 75 %; Placebo: 0 % bzw. 13 %).
Pädiatrische Plaque-Psoriasis
Eine randomisierte, doppelblinde, multizentrische, placebokontrollierte Studie (IXORA-Peds) umfasste 201 pädiatrische Patienten im Alter von 6 bis unter 18 Jahren mit mittelschwerer bis schwerer Plaque-Psoriasis (definiert durch einen sPGA-Wert ≥3, der ≥10% der Körperoberfläche einbezieht, und einen PASI-Wert ≥12), die Kandidaten für eine Phototherapie oder systemische Therapie waren oder bei einer topischen Therapie unzureichend kontrolliert waren.
Die wesentliche Ausschlusskriterien waren ähnlich wie bei den oben genannten Studien in Erwachsenen, insbesondere aktive oder kürzlich durchgemachte schwerwiegende Infektionen sowie begleitende systemische konventionelle oder biologische Psoriasis-Therapien oder Phototherapie.
Während der 12-wöchigen doppelblinden Placebo- und aktiv-kontrollierten Phase wurden die Patienten mit Placebo (n=56), Etanercept (n=30) oder Taltz (n=115) behandelt, wobei dieGewichts-adaptierte Dosierung wie folgt angepasst wurde:
Taltz: <25 kg: 40 mg in Woche 0, danach 20 mg Q4W
25 kg bis 50 kg: 80 mg in Woche 0, danach 40 mg Q4W
>50 kg: 160 mg in Woche 0, danach 80 mg Q4W
Etanecerpt:0.8 mg/kg, maximal 50 mg pro Dosis jede Woche.
Das Ansprechen auf die Behandlung wurde nach 12 Wochen Therapie beurteilt und wurde durch den Anteil der Patienten definiert, die den co‑primären Endpunkt eines sPGA-Wert von «0» (frei von) oder «1» (minimal) mit einer Verbesserung von mindestens 2 Punkten gegenüber dem Ausgangswert (Baseline) erreichten, und den Anteil der Patienten, die eine Reduktion des PASI-Wertes von mindestens 75% (PASI 75) gegenüber Baseline erreichten.
Andere evaluierte Ergebnisse in Woche 12 umfassten den Anteil der Patienten, die PASI 90, PASI 100, sPGA von «0» erreichten, und eine Verbesserung des Schweregrads des Juckreizes erreichten, gemessen durch eine Reduktion von mindestens 4 Punkten auf einer 11-Punkte-Juckreiz-Bewertungsskala (itch Numeric Rating Scale, itch NRS).
Die Patienten hatten einen mittleren Baseline PASI-Wert von 17, mit Werten zwischen 12 und 49. Der Baseline sPGA-Wert lag bei 49% der Patienten bei schwer oder sehr schwer. Von allen Patienten erhielten 22% eine vorherige Phototherapie, 32% eine vorherige konventionelle systemische Therapie und 4% eine Biologika-Vortherapie zur Behandlung der Psoriasis.
Die Daten des klinischen Ansprechens sind in Tabelle 4 aufgeführt.
Tabelle 4: Ergebnisse der Wirksamkeit nach 12 Wochen bei pädiatrischen Patienten mit Plaque Psoriasis, NRI
Endpunkte | Taltza (N=115) n (%) | Placebo (N=56) n (%) | Unterschied zu Placebo (95% CI) |
|---|---|---|---|
sPGA «0» (frei von) oder «1» (minimal)b | 93 (81) | 6 (11) | 70.2 (59.3, 81.0) d |
sPGA «0» (frei von) | 60 (52) | 1 (2) | 50.4 (40.6, 60.2) d |
PASI 75c | 102 (89) | 14 (25) | 63.7 (51.0, 76.4)d |
PASI 90 | 90 (78) | 3 (5) | 72.9 (63.3, 82.5)d |
PASI 100 | 57 (50) | 1 (2) | 47.8 (38.0, 57.6)d |
Itch NRS (≥4 Punkte Verbesserung) c | 59 (71) | 8 (20) | 51.1 (35.3, 66.9) d |
Abkürzungen: N =Anzahl Patienten in der Intent-to-treat-Population; NRI = Non-responder Imputation.
a In Woche 0 erhielten die Patienten 160 mg (Körpergewicht [KG] >50kg), 80 mg (KG 25-50 kg), oder 40 mg (KG < 25 kg) Taltz, danach 80 mg (KG >50kg), 40 mg (KG 25-50 kg) oder 20 mg (KG < 25 kg) alle 4 Wochen, für 12 Wochen.
b Co-primäre Endpunkte.
c Itch NRS (≥4 Verbesserung) bei Patienten mit Baseline itch NRS ≥4. Anzahl der ITT-Patienten mit Baseline itch NRS-Wert ≥4 wie folgt: Taltz n = 83; Placebo n = 40.
d p<0.001
Insgesamt wurden 87 pädiatrische Patienten mit schwerer Plaque-Psoriasis (PASI ≥20 oder sPGA ≥4) zu Ixekizumab Q4W (38 Patienten), Etanercept Q1W (30 Patienten) oder Placebo (19 Patienten) randomisiert.
In Woche 12 wurden Verbesserungen für die Ixekizumab Q4W-Gruppe im Vergleich zur Etanercept Q1W-Gruppe und zur Placebo-Gruppe beobachtet, gemessen mit PASI 75 (84.2%, 63.3%, 26.3%) und sPGA (0,1) (76.3%, 53.3% und 5.3%).
In den wichtigsten sekundären Endpunkte zeigten sich statistisch signifikante Verbesserungen in der Ixekizumab Q4W-Gruppe im Vergleich zur Etanercept Q1W-Gruppe und zur Placebo-Gruppe, gemessen an: PASI 90 (76.3%, 40.0%, 0), PASI 100 (60.5%, 16.7%, 0) und sPGA (0) (63.2%, 16.7%, 0 )
Patienten in der Ixekizumab-Behandlungsgruppe hatten ein höheres CDLQI (children Dermatology Life Quality Index)/DLQI (0,1) Ansprechen in Woche 12 (NRI) im Vergleich zu Placebo. Der Unterschied zwischen den Behandlungsgruppen zeigte sich bereits ab Woche 4.
Es liegen keine Daten nach Absetzen von Taltz bei Kindern und zum Risiko eines Aufflammens der Krankheit vor.
Psoriasis-Arthritis
Die Sicherheit und Wirksamkeit von Taltz wurden an 780 Patienten in zwei randomisierten, doppelblinden placebo-kontrollierten Phase III Studien bei Patienten mit aktiver Psoriasis-Arthritis (≥3 geschwollene und ≥3 schmerzhafte Gelenke) untersucht. Bei den Patienten in diesen Studien wurde die Diagnose einer Psoriasis Arthritis (CASPAR Klassifizierungskriterien, Classification Criteria for Psoriatic Arthritis) im Median 5.33 Jahren vor Studieneinschluss gestellt. Randomisierte Patienten hatten gleichzeitig auch Plaque-Psoriasis Hautläsionen (94.0 %) oder eine dokumentierte Plaque-Psoriasis in der Anamnese. Bei Studienbeginn litten 12.1 % der Patienten an einer mittelschweren bis schweren Plaque-Psoriasis. Von den Psoriasis-Arthritis Patienten hatten zu Studienbeginn mehr als 58.9 % eine Enthesitis bzw. 22.3 % eine Daktylitis. Für beide Studien war der primäre Endpunkt das American College of Rheumatology (ACR) 20 Ansprechen in Woche 24.
In der Psoriasis-Arthritis Studie 1 (SPIRIT-P1), wurden Biologika -naive Patienten mit aktiver Psoriasis-Arthritis in folgende Therapiegruppen randomisiert: subkutane Injektionen mit Placebo, Adalimumab 40 mg alle 2 Wochen (aktive Kontrollgruppe), Taltz 80 mg alle 2 Wochen (Q2W), oder 80 mg alle 4 Wochen (Q4W). Beide Taltz-Dosierschema beinhalteten eine Anfangsdosis von 160 mg. 85.3% der Patienten in dieser Studie hatten eine vorherige Behandlung mit ≥1 cDMARD erhalten. 53% der Patienten haben gleichzeitig MTX mit einer mittleren wöchentlichen Dosis von 15.8 mg angewendet. 67% der Patienten, die gleichzeitig mit MTX behandelt wurden, hatten eine Dosis von 15 mg oder mehr. Patienten in allen Behandlungsgruppen mit einem unzureichenden Ansprechen in Woche 16 erhielten eine Rettungstherapie (Modifikation der Hintergrundtherapie). Patienten auf Taltz Q2W oder Q4W blieben auf deren initial bestimmter Taltz Dosis. Patienten, die Adalimumab oder Placebo erhielten, wurden in Woche 16 oder 24 basierend auf den Responderstatus 1:1 auf Taltz Q2W oder Q4W neu randomisiert.
Psoriasis-Arthritis Studie 2 (SPIRIT-P2) schloss Patienten ein, die zuvor mit einem anti-TNF Wirkstoff behandelt wurden und den anti-TNF Wirkstoff wegen ungenügender Wirksamkeit oder Unverträglichkeit (anti-TNF-IR Patienten) abgebrochen haben. Patienten wurden in folgende Therapiegruppen randomisiert: subkutane Injektionen mit Placebo, Taltz 80 mg alle 2 Wochen (Q2W), oder 80 mg alle 4 Wochen (Q4W). Beide Taltz-Dosierschema beinhalteten eine Anfangsdosis von 160 mg. 56% und 35% der Patienten haben unzureichend auf 1 TNF bzw auf ≥2 TNF angesprochen. In SPIRIT-P2 wurden 363 Patienten ausgewertet, von denen 41% gleichzeitig MTX bei einer mittleren wöchentlichen Dosis von 16.1 mg angewendet haben. 73.2% der Patienten, die gleichzeitig mit MTX behandelt wurden, hatten eine Dosis von 15 mg oder mehr. Patienten in allen Behandlungsgruppen mit einem unzureichendem Ansprechen in Woche 16 erhielten eine Rettungstherapie (Modifikation der Hintergrundtherapie). Patienten auf Taltz Q2W oder Q4W blieben auf deren initial bestimmter Taltz Dosis. Patienten, die Placebo erhielten, wurden in der Woche 16 oder 24 basierend auf den Responderstatus 1: 1 auf Taltz Q2W oder Q4W neu randomisiert.
Anzeichen und Symptome
Die Behandlung mit Taltz zeigte in der Woche 24 eine signifikante Verbesserung der Krankheitsaktivität im Vergleich zu Placebo (siehe Tabelle 5).
Tabelle 5. Wirksamkeitsergebnisse in SPIRIT-P1 und SPIRIT-P2 in Woche 24
SPIRIT-P1 | SPIRIT-P2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Endpunkte | Unterschied von Placebo in Ansprechrate (95% CI) | Unterschied von Placebo in Ansprechrate (95% CI) | ||||||||
PBO (N = 106) | Taltz Q4W (N = 107) | Taltz Q2W (N = 103) | Taltz Q4W | Taltz Q2W | PBO (N = 118) | Taltz Q4W (N = 122) | Taltz Q2W (N = 123) | Taltz Q4W | Taltz Q2W | |
ACR 20 Ansprechen, n (%) | ||||||||||
Woche 24 | 32 (30.2) | 62 (57.9) | 64 (62.1) | 27.8 (15.0, 40.6)* | 31.9 (19.1, 44.8) * | 23 (19.5) | 65 (53.3) | 59 (48.0) | 33.8 (22.4, 45.2) * | 28.5 (17.1, 39.8) * |
ACR 50 Ansprechen, n (%) | ||||||||||
Woche 24 | 16 (15.1) | 43 (40.2) | 48 (46.6) | 25.1 (13.6, 36.6) * | 31.5 (19.7, 43.3) * | 6 (5.1) | 43 (35.2) | 41 (33.3) | 30.2 (20.8, 39.5) * | 28.3 (19.0, 37.5) * |
ACR 70 Ansprechen, n (%) | ||||||||||
Woche 24 | 6 (5.7) | 25 (23.4) | 35 (34.0) | 17.7 (8.6, 26.8) * | 28.3 (18.2, 38.5) * | 0 | 27 (22.1) | 15 (12.2) | 22.1 (14.8, 29.5) * | 12.2 (6.4, 18.0) * |
Abkürzungen: ACR 20/50/70 = American College of Rheumatology 20%/50%/70% Ansprechrate; CI = confidence interval; Q4W = Taltz 80 mg alle 4 Wochen; Q2W = Taltz 80 mg alle 2 Wochen; N = Anzahl der Patienten in der untersuchten Population; n = Anzahl der Patienten in der spezifischen Kategorie; NRI = non-responder imputation; PBO = Placebo.
Hinweis: Patienten die in Woche 16 die Rettungstherapie bekommen haben oder die Therapie abgebrochen hatten, oder bei denen es fehlende Daten gab, wurden als Non-Responder für die Analysen der Woche 24 gezählt.
Gleichzeitige cDMARDs Anwendung schloss MTX, Leflunomid und Sulfasalazin ein.
* p<0.001 im Vergleich zu Placebo.
Bei Patienten mit bereits vorhandener Daktylitis oder Enthesitis führte die Behandlung mit Taltz Q4W zu einer Verbesserung der Daktylitis bzw. der Enthesitis in Woche 24 im Vergleich zu Placebo (p<0.01).
Bei Patienten mit gleichzeitiger Plaque-Psoriasis (≥3% BSA) und Psoriasis-Arthritis führte die Behandlung mit Taltz Q4W zu einer Verbesserung psoriatischer Hautläsionen, wie anhand von PASI 75-, PASI 90- und PASI 100-Ansprechen in Woche 24 gezeigt werden konnte (p < 0.001).
Bei Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis und Psoriasis-Arthritis zeigte Taltz Q2W Dosierungsschema signifikante höhere Ansprechrate für PASI 75, PASI 90 und PASI 100 im Vergleich zu Placebo (p<0.001) und einen klinisch bedeutsamen Nutzen gegenüber dem Q4W Dosierungsschema.
Das Ansprechen auf die Behandlung mit Taltz war bereits in Woche 1 für ACR 20, in Woche 4 für ACR 50 signifikant höher als das Ansprechen auf Placebo und in Woche 8 für ACR 70 und wurde durchgängig bis Woche 24 beibehalten.
Abbildung 2. ACR 20 Ansprechen in SPIRIT-P1 über die Zeit bis zur Woche 24

Für Taltz Q2W und Q4W: b p<0.01 und c p<0.001 im Vergleich zu Placebo.
In SPIRIT-P1 und SPIRIT-P2 wurden ähnliche Ansprechen für ACR 20/50/70 in Psoriasis-Arthritis Patienten beobachtet, ungeachtet der Tatsache ob eine Begleittherapie mit cDMARDs, einschliesslich MTX, vorhanden war oder nicht.
In SPIRIT-P1 und SPIRIT-P2 konnten in allen Komponenten der ACR Scores, einschliesslich der Patientenbeurteilung zum Schmerz, Verbesserungen gezeigt werden.
In SPIRIT-P1 wurde anhaltend die Wirksamkeit über 52 Wochen anhand der Ansprechraten auf die Parameter ACR 20/50/70, Besserung der Enthesitis, Besserung der Dactylitis und PASI 75/90 /100, gezeigt.
Die Wirksamkeit und Sicherheit von Taltz wurde ungeachtet von Alter, Geschlecht, Abstammung, Dauer der Erkrankung, Körpergewicht zu Studienbeginn, Psoriasis Beteiligung zu Studienbeginn, CRP Ausgangwert, DAS28-CRP Ausgangwert, Begleittherapie mit Kortikosteroiden, und vorherige Behandlung mit Biologika gezeigt. Taltz war wirksam bei Biologika-naiven, Biologika-exponierten und Biologika Non-Responder Patienten.
Es gab zu wenige Patienten mit Arthritis mutilans, isolierter Arthritis mit Befall der distalen Interphalangealgelenken (DIP) und begleitender Spondylitis in den Zulassungsstudien, um aussagekräftige Resultate in diesen spezifischen Untergruppen zu erhalten.
Radiographisches Ansprechen
In SPIRIT-P1 wurde die Hemmung der Progression struktureller Gelenkschäden radiographisch untersucht, und anhand von modifiziertem Total Sharp Score (mTSS) und seinen Komponenten Erosions Score (ES) und dem Joint Space Narrowing Score (JSN) in den Wochen 24 und 52 im Vergleich zu den Ausgangswerten gemessen.
Taltz hemmte die Progression struktureller Gelenkschäden (mTSS) im Vergleich zu Placebo in Woche 24.
Der Prozentanteil der Patienten ohne radiographische Progression struktureller Gelenkschäden (definiert als eine mTSS Veränderung gegen dem Ausgangswert ≤0.5) von der Randomisierung bis zur Woche 24 betrug 94.8% für Taltz Q2W, 89.0 % für Taltz Q4W, und 77.4% für Placebo. Die Hemmung der strukturellen Schäden wurde mit der Taltz-Behandlung bis zur Woche 52 beibehalten.
Körperliche Funktion und gesundheitsrelevante Lebensqualität
In SPIRIT-P1 und SPIRIT-P2 zeigten mit Taltz Q2W (p<0.001) und Q4W (p<0.001) behandelte Patienten eine durch Health Assesment Questionnaire-Disability Index (HAQ-DI) bewertet Verbesserung der körperlichen Funktion in Woche 24. Diese wurde in SPIRIT-P1 bis Woche 52 beibehalten.
Patienten, die mit Taltz behandelt wurden, berichteten von Verbesserungen in der gesundheitsrelevanten Lebensqualität, die mithilfe des Physical Component Summary des Short Form‑36 Health Survey (SF‑36 PCS) Score gemessen wurden (p < 0.001). Auch eine statistisch signifikante Verbesserung der Müdigkeit konnte im Fatigue Severity NRS gezeigt werden.
Ankylosierende Spondylitis (radiographische axiale Spondyloarthritis, Morbus Bechterew)
Die Sicherheit und Wirksamkeit von Taltz wurden bei 657 Patienten in 2 randomisierten, doppelblinden, placebokontrollierten Studien (COAST-V und COAST-W) bei erwachsenen Patienten ≥18 Jahren mit ankylosierender Spondylitis untersucht. Die Patienten hatten eine aktive Krankheit definiert nach dem Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 und trotz einer Therapie mit NSAID Wirbelsäulenschmerzen ≥4 auf einer numerischen Bewertungsskala. In beiden Studien hatten die Patienten bei Baseline Symptome einer ankylosierenden Spondylitis über einen Zeitraum von durchschnittlich 17 Jahren (Median von 16 Jahren). Bei Baseline erhielten etwa 32% der Patienten begleitend eine cDMARD-Therapie (in COAST-V und in COAST-W: Methotrexat 8.5% und 13.0%, Sulfasalazin 28.5% und 14.6%, Hydroxychloroquin 0.6% und 0%).
Primärer Endpunkt in beiden Studien war der Prozentsatz von Patienten, die in Woche 16 ein Assessment of Spondyloarthritis International Society 40 (ASAS40)-Ansprechen erreichten.
COAST-V bewertete 341 biologisch-naive Patienten, die entweder mit Taltz 80 mg oder 160 mg in Woche 0, gefolgt von 80 mg alle 2 Wochen (Q2W) oder 4 Wochen (Q4W), Adalimumab 40 mg alle 2 Wochen oder mit Placebo behandelt wurden. Die mit Placebo behandelten Patienten wurden in Woche 16 neu randomisiert, um Taltz zu erhalten (160 mg Startdosis, gefolgt von 80 mg Q2W oder Q4W). Die mit Adalimumab behandelten Patienten wurden in Woche 16 neu randomisiert, um Taltz zu erhalten (80 mg Q2W oder Q4W).
COAST-W bewertete 316 Patienten, die Vorerfahrungen mit 1 oder 2 TNF-Hemmern hatten (90% hatten ein unzureichendes Ansprechen, und 10% hatten eine Unverträglichkeit auf TNF-Hemmer). Alle Patienten wurden mit Taltz 80 oder 160 mg in Woche 0 und danach mit 80 mg Q2W oder Q4W oder mit Placebo behandelt. Die mit Placebo behandelten Patienten wurden in Woche 16 neu randomisiert, um Taltz zu erhalten (160 mg Startdosis, gefolgt von 80 mg Q2W oder Q4W).
Klinisches Ansprechen
In beiden Studien wiesen die mit Taltz 80 mg Q2W oder 80 mg Q4W behandelten Patienten in Woche 16 grössere Verbesserungen im ASAS40- und ASAS20-Ansprechen im Vergleich zu Placebo auf (Tabelle 6). Das Ansprechen war bei den Patienten unabhängig von den begleitenden Therapien vergleichbar. Bei COAST-W zeigte sich das Ansprechen unabhängig von der Anzahl der früheren Therapien mit TNF-Hemmern.
Tabelle 6. Wirksamkeitsergebnisse in COAST-V und COAST-W in Woche 16
COAST-V, Biologika-naiv | COAST-W, TNF-Hemmer-erfahren | ||||||
|---|---|---|---|---|---|---|---|
Taltz 80 mg Q4Wa (N=81) | Placebo (N=87) | Unterschied zu Placebo (95% CI) | Adalimumab 40 mg Q2W (N= 90) | Taltz 80 mg Q4Wc (N=114) | Placebo (N=104) | Unterschied zu Placebo (95% CI) | |
ASAS20-Ansprechenb, n (%), NRI | 52 (64.2%) | 35 (40.2%) | 24.0 (9.3, 38.6)** | 53 (58.9%) | 55 (48.2%) | 31 (29.8%) | 18.4 (5.7, 31.1)** |
ASAS40-Ansprechenb,c, n (%), NRI | 39 (48.1%) | 16 (18.4%) | 29.8 (16.2, 43.3)*** | 32 (35.6%) | 29 (25.4%) | 13 (12.5%) | 12.9 (2.7, 23.2)* |
ASDAS Baseline [CFB] | 3.7 | 3.9 | -1.0 (-1.3, -0.7)*** | 3.7 | 4.2 [‑1.2] | 4.1 | -1.1 (-1.3, -0.8)*** |
BASDAI-Score Baseline [CFB] | 6.8 [-2.9] | 6.8 [-1.4] | -1.5 (-2.1, -0.9)*** | 6.7 [-2.5]*** | 7.5 [‑2.2] | 7.3 [-0.9] | -1.2 (-1.8, -0.7)*** |
MRI Spine SPARCCd Baseline [CFB] | 14.5 [‑11.0] | 15.8 | -9.5 (-12.6, -6.4)*** | 20.0 | 8.3 [‑3.0] | 6.4 | -6.3 (-10.0, -2.5)** |
BASDAI50e (%), NRI | 42 | 17 | 25 (11,38)*** | 32* | 22 | 10 | 12 (3, 22)* |
ASDAS <2.1 (%) (Geringe Krankheitsaktivität), NRI | 43 | 13 | 31 (18,43)*** | 38*** | 18 | 5 | 13 (5, 21)** |
ASAS HI Baseline [CFB] | 7.5 | 8.1 | -1.1 (-2.0, -0.3)* | 8.2 | 10.0 [‑1.9] | 9.0 | -1.0 (-1.9, -0.1)* |
SF-36 PCS Baseline [CFB] | 34.0 [7.7] | 32.0 [3.6] | 4.1 (1.9, 6.2)*** | 33.5 [6.9]** | 27.5 [6.6]*** | 30.6 [1.4] | 5.2 (3.0, 7.4)*** |
Abkürzungen: N = Anzahl Patienten in der Intent-to-treat-Population; NRI = Non-responder Imputation; Patienten mit fehlenden Daten wurden als Non-Responder gewertet.
ASAS HI = Assessment of SpondyloArthritis International Society Health Index; ASDAS = Ankylosing Spondylitis Disease Activity Score; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; CFB = least square mean change from baseline in Woche 16; MRI Spine SPARCC = Spondyloarthritis Research Consortium of Canada Magnetic Resonance Imaging Scoring of the Spine (23 discovertebral unit scale), SF-36 PCS = Short Form health survey physical component summary.
a In Woche 0 erhielten Patienten 80 mg oder 160 mg Taltz.
b ASAS20-Ansprechen: Verbesserung um ≥20% und absolute Verbesserung um ≥1 Einheit (Skala von 0–10) gegenüber Baseline bei ≥3 von 4 Bereichen (Patient Global, Spinal Pain, Function, Inflammation), wobei keine Verschlechterung des verbleibenden Bereichs (definiert als Verschlechterung um ≥20% und durchschnittliche Verschlechterung um ≥1 Einheit, Skala von 0–10) zu verzeichnen ist. ASAS40-Ansprechen: Verbesserung um ≥40% und absolute Verbesserung gegenüber Baseline um ≥2 Einheiten in ≥3 von 4 Bereichen ohne Verschlechterung im verbleibenden Bereich.
c Primärer Endpunkt.
d Die Anzahl der ITT-Patienten mit MRT-Daten zu Studienbeginn lautet: COAST-V: Taltz, n = 81; PBO, n = 82, ADA, n=85. COAST-W: Taltz, n = 58; PBO, n = 51.
e BASDAI50-Ansprechen ist definiert als eine Verbesserung von ≥50% des BASDAI-Score gegenüber dem Ausgangswert.
* p<0.05; ** p<0.01; *** p<0.001 im Vergleich zu Placebo.
Die Verbesserung der Hauptkomponenten der ASAS40-Ansprechkriterien (Wirbelsäulenschmerzen, BASFI, globale Beurteilung des Patienten, Steifheit) und anderer Messungen der Krankheitsaktivität, einschliesslich CRP, zeigte in Woche 16 eine signifikante klinische Verbesserung.
Der Prozentsatz der Patienten, die bei Untersuchungen in COAST-V und COAST-W ein ASAS40-Ansprechen erreichten, ist in Abbildung 3 dargestellt.
Abbildung 3. ASAS40-Ansprechen in COAST-V und COAST-W bis Woche 16, NRIa

a Patienten mit fehlenden Daten wurden als Non-Responder gewertet.
* p<0.05; ** p<0.01; *** p<0.001 im Vergleich zu Placebo.
Vergleichbare ASAS40-Ansprechraten wurden bei Patienten unabhängig von CRP zu Studienbeginn, von ASDAS-Scores zu Studienbeginn und von MRI Spine SPARCC-Scores festgestellt. Der ASAS40 Ansprechen wurde unabhängig von Alter, Geschlecht, Rasse, Krankheitsdauer, Körpergewicht zu Studienbeginn, BASDAI-Score zu Studienbeginn und vorheriger biologischer Behandlung nachgewiesen.
In COAST-V und COAST-W wurde die Wirksamkeit bis zu Woche 52 beibehalten, wie dies durch die oben in Woche 16, dargestellten Endpunkte, einschliesslich ASAS20-, ASAS40-, ASDAS-, BASDAI-, BASFI- und ASAS HI-Ansprechen beurteilt wurde.
Gesundheitsbezogene Ergebnisse
Es zeigten sich bereits ab Woche 1 Verbesserungen bei Rückenschmerzen gegenüber Placebo, und diese wurden bis Woche 16 aufrechterhalten (Taltz vs. Placebo: COAST-V -3.2 vs. -1.7; COAST-W -2.4 vs. -1.0); Müdigkeit und Wirbelsäulenbeweglichkeit zeigten in Woche 16 Verbesserungen gegenüber Placebo. Die Verbesserungen von Rückenschmerzen, Müdigkeit und Wirbelsäulenbeweglichkeit blieben bis zu Woche 52 erhalten.
Pharmakokinetik
Die pharmakokinetische Eigenschaften von Taltz waren bei den Indikationen Plaque-Psoriasis, Psoriasis-Arthritis, und ankylosierende Spondylitis vergleichbar.
Absorption
Bei Patienten mit Psoriasis wurden nach subkutaner (SC) Gabe einer Ixekizumab Einzeldosis im Dosisbereich von 5 bis 160 mg innerhalb von 4 bis 7 Tagen mittlere maximale Serumkonzentrationen erreicht. Die mittlere (Standardabweichung) maximale Serumkonzentration (Cmax) von Ixekizumab nach einer Startdosis von 160 mg betrug 19.9 (8.15) µg/ml.
Nach einer Startdosis von 160 mg wurde der Steady-State mit dem 80 mg Q2W Dosierungsschema in der Woche 8 erreicht. Die mittleren (Standardabweichung) geschätzten Cmax,ss, und C trough,ss betrugen 21.5 (9.16) µg/ml und 5.23 (3.19) µg/ml.
Nach Umstellung der Dosierungsschema in Woche 12 von 80 mg Q2W auf 80 mg Q4W, würde der Steady-State nach etwa 10 Wochen erreicht werden. Die mittleren (Standardabweichung) geschätzten Cmax,ss, und C trough,ss betrugen 14.6 (6.04) µg/ml und 1.87 (1.30) µg/ml.
Die durchschnittliche absolute Bioverfügbarkeit von Ixekizumab lag über die Analysen hinweg geschätzt in einem Bereich von 54 % bis 90 %. Nach subkutaner Injektion in den Bauch, Oberarm oder Oberschenkel werden ähnliche Ixekizumab Serumkonzentrationen erreicht.
Distribution
Basierend auf den Populations-pharmakokinetischen Analysen betrug das mittlere gesamte Verteilungsvolumen im Steady-State 7.11 Liter.
Metabolismus
Ixekizumab ist ein monoklonaler Antikörper, sodass in gleicher Art wie bei endogenen IgGs ein Abbau über katabole Prozesse in kleine Peptide und Aminosäuren erwartet wird.
Elimination
In der Populations-pharmakokinetischen Auswertung betrug die mittlere Serum-Clearance 0.0161 l/Std. Die Clearance ist von der Dosis unabhängig. Basierend auf Schätzungen der Populations-pharmakokinetischen Auswertung betrug die mittlere Eliminations-Halbwertszeit bei Patienten mit Plaque Psoriasis 13 Tage.
Linearität/Nicht Linearität
Die Exposition (AUC) im Dosisbereich von 5 bis 160 mg als subkutane Injektion stieg proportional zur verabreichten Dosis an.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Spezifische klinische pharmakologische Studien zur Untersuchung der Auswirkungen von Leberfunktionsinsuffizienz auf die Pharmakokinetik von Ixekizumab wurden nicht durchgeführt.
Nierenfunktionsstörungen
Spezifische klinische pharmakologische Studien zur Untersuchung der Auswirkungen von Nierenfunktionsinsuffizienz auf die Pharmakokinetik von Ixekizumab wurden nicht durchgeführt.
Ältere Patienten
Basierend auf einer populationspharmakokinetischen Analyse mit einer begrenzten Anzahl älterer Patienten (n = 94 mit ≥65 Jahre und n = 12 mit ≥75 Jahre) war die Clearance bei älteren Patienten mit der Clearance bei Patienten unter 65 Jahren vergleichbar.
Kinder und Jugendliche
Pädiatrische Psoriasis-Patienten (Alter 6 bis weniger als 18 Jahre) wurden 12 Wochen lang mit Ixekizumab im empfohlenen pädiatrischen Dosierungsschema behandelt. Patienten mit einem Gewicht von >50 kg und 25 bis 50 kg hatten in Woche 12 eine mittlere ±SD Steady-state-Talspiegelkonzentration von 3.8 ± 2.2 µg/ml bzw. 3.9 ± 2.4 µg/ml. Diese Konzentrationen waren vergleichbar mit Konzentrationen bei erwachsenen Patienten, die mit Ixekizumab 80 mg Q4W dosiert wurden, wobei die mittlere ±SD Steady-state-Talspiegelkonzentration 3.5 ± 2.2 μg / ml betrug.
Präklinische Daten
Nicht-klinische Daten von Cynomolgus Affen zeigten basierend auf Studien zur Toxizität bei wiederholter Gabe, pharmakologischen Auswertungen zur Sicherheit, sowie Studien zur Entwicklungs- und Reproduktionstoxizität keine besonderen Gefahren für den Menschen.
Toxizität bei wiederholter Verabreichung
Bei Cynomolgus Affen rief die subkutane Gabe von Ixekizumab in Dosierungen von bis zu 50 mg/kg/Woche über 39 Wochen keine Organtoxizität oder unerwünschten Wirkungen auf die Immunfunktion hervor (z.B. T-Zell-abhängige Antikörper-Antworten und NK-Zell-Aktivität). Die wöchentliche subkutane Gabe von 50 mg/kg bei Affen entspricht etwa dem 19-Fachen der Taltz 160 mg Startdosis und führt bei Affen zu einer Exposition (AUC) die mindestens 61-fach höher ist als die mittlere vorhergesagte Exposition beim Menschen im Steady-State bei empfohlener Dosierung.
Mutagenität / Karzinogenität
Nicht-klinische Studien zur Untersuchung des karzinogenen oder mutagenen Potentials von Ixekizumab wurden nicht durchgeführt.
Reproduktionstoxizität
Bei geschlechtsreifen Cynomolgus Affen, die über 13 Wochen hinweg wöchentlich 50 mg/kg Ixekizumab subkutan erhalten haben, wurden keine Wirkungen auf die Reproduktionsorgane, den Menstruationszyklus oder die Spermien beobachtet.
In Studien zur Entwicklungstoxizität führte die subkutane Gabe von Ixekizumab in Dosierungen bis zu 50 mg/kg einmal wöchentlich bei Cynomolgus Affen vom Beginn der Organogenese bis entweder kurz vor Ende der Schwangerschaft oder bis zur Geburt nicht zu Embryotoxizität oder Teratogenität und hatte keine Wirkungen auf den Geburtsverlauf oder die morphologische, funktionelle oder immunologische Entwicklung des Nachwuchs von der Geburt bis zu einem Alter von 6 Monaten. Die höhere Inzidenz von postnatalen Todesfällen beim Affen war in erster Linie auf mütterliche Vernachlässigung oder Frühgeburten zurückzuführen. Es handelt sich dabei um Befunde, die beim Affen häufig auftreten und vermutlich nicht auf Ixekizumab zurückzuführen sind. Wegen der geringen Tierzahlen ist der Vorhersagewert der Studie begrenzt. Es wurde gezeigt, dass Ixekizumab plazentagängig ist und im Blut des Nachwuchses bis zu einem Alter von 6 Monaten vorhanden war.
Sonstige Hinweise
Inkompatibilitäten
Nicht zutreffend (einzeldosiertes Arzneimittel zur subkutanen Anwendung)
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8°C) lagern. In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Nicht einfrieren. Taltz, das eingefroren war, darf nicht verwendet werden.
Temporäre Lagerung: Taltz kann bis zu 5 Tagen ungekühlt bei Temperaturen unter 30°C aufbewahrt werden.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Nicht schütteln.
Taltz ist eine sterile, Konservierungsmittel-freie, klare und farblose bis leicht gelbliche Lösung.
Nicht verwenden, wenn Partikel auftreten oder wenn die Lösung wolkig und/oder merklich braun ist.
Die Fertigspritze und der Fertigpen sind zum einmaligen Gebrauch bestimmt.
Die der Patienteninformation beigelegte Bedienungsanleitung der Fertigspritze bzw. des Fertigpens muss sorgfältig beachtet werden.
Vorbereitung von Taltz 40 mg für pädiatrische Patienten von 25 kg bis zu 50 kg
Ixekizumab-Dosen von 40 mg müssen von einer qualifizierten medizinischen Fachperson vorbereitet und verabreicht werden. Verwenden Sie nur die im Handel erhältliche Taltz 80 mg/1 ml-Fertigspritze, wenn Sie die vorgeschriebene pädiatrische Dose von 40 mg vorbereiten.
- den gesamten Inhalt der Fertigspritze in eine sterile Durchstechflasche aus klarem Glas injizieren. Die Durchstechflasche NICHT schütteln oder schwenken.
- eine 0.5-ml- oder 1-ml-Einwegspritze und eine sterile Nadel verwenden, um 0.5 ml aus der Durchstechflasche zu entnehmen.
- die Nadel wechseln und eine 27 Gauge sterile Nadel verwenden, um dem Patienten die Injektion zu verabreichen. Unbenutztes Ixekizumab in der Durchstechflasche entsorgen.
Das vorbereitete Ixekizumab muss innerhalb von 4 Stunden nach dem Durchstechen der sterilen Durchstechflasche bei Raumtemperatur verabreicht werden.
Zulassungsnummer
65906, 65907 (Swissmedic).
Zulassungsinhaberin
Eli Lilly (Suisse) SA, 1214 Vernier/GE.
Stand der Information
Juli 2020
Composizione
Principi attivi
Ixekizumab (da cellule CHO (cellule ovariche di criceto cinese) ottenute mediante ingegneria genetica).
Sostanze ausiliarie
Citrato di sodio diidrato, acido citrico, cloruro di sodio, polisorbato 80, acqua per preparazioni iniettabili q.s. ad solutionem pro 1 ml.
Quantità totale di sodio: 5.8 mg/ml.
Forma farmaceutica e quantità di principio attivo per unità
Taltz 80 mg soluzione iniettabile in siringa preriempita
Ogni siringa preriempita contiene 80 mg di ixekizumab in 1 ml (80 mg/ml).
Taltz 80 mg soluzione iniettabile in penna preriempita
Ogni penna preriempita contiene 80 mg di ixekizumab in 1 ml (80 mg/ml).
Indicazioni/Possibilità d'impiego
Psoriasi a placche
Taltz è indicato per il trattamento della psoriasi a placche di grado da moderato a severo in adulti che non hanno reagito ad altre terapie sistemiche (fra cui ciclosporina o metotressato o PUVA), per i quali queste terapie sono controindicate o che sono intolleranti a queste terapie.
Psoriasi a placche in bambini e adolescenti
Taltz è indicato per il trattamento della psoriasi a placche di grado da moderato a severo in bambini e adolescenti dai 6 anni e con un peso corporeo di almeno 25 kg, che non hanno reagito ad altre terapie sistemiche (fra cui ciclosporina o metotrexato o PUVA), per i quali queste terapie sono controindicate o che sono intolleranti a queste terapie.
Artrite psoriasica
Taltz, da solo o in associazione a medicamenti antireumatici modificanti la malattia (DMARD, disease-modifying anti-rheumatic drugs), è indicato per il trattamento dell'artrite psoriasica attiva in pazienti adulti che hanno avuto una risposta insufficiente al trattamento con uno o più medicamenti DMARD o che non lo hanno tollerato.
Spondiloartrite assiale
- Spondilite anchilosante (morbo di Bechterew)
Taltz è indicato per il trattamento della spondilite anchilosante attiva severa in pazienti adulti che hanno avuto una risposta insufficiente a una terapia convenzionale (ad es. antinfiammatori non steroidei [FANS]) o che non la tollerano.
- Spondiloartrite assiale non radiografica
Taltz è indicato per il trattamento della spondiloartrite assiale non radiografica attiva severa in pazienti adulti che hanno avuto una risposta insufficiente ai FANS. I pazienti devono presentare segni obiettivi di infiammazione nella tomografia a risonanza magnetica (MRT) e tramite valori elevati di proteina C-reattiva (CRP).
Posologia/Impiego
Taltz deve essere usato sotto la guida e la supervisione di un medico con esperienza nella diagnosi e nel trattamento delle patologie per le quali Taltz è indicato.
Al fine di garantire la tracciabilità dei medicamenti di produzione biotecnologica, il nome commerciale e il numero di lotto del prodotto somministrato devono essere documentati a ogni trattamento.
Posologia abituale
Psoriasi a placche negli adulti
La dose raccomandata è di 160 mg somministrata per via sottocutanea (due iniezioni da 80 mg) alla settimana 0, seguita da una dose di 80 mg (una iniezione) alle settimane 2, 4, 6, 8, 10 e 12, e poi da una dose di 80 mg (una iniezione) ogni 4 settimane.
Nei pazienti <100 kg può essere preso in considerazione uno schema di dosaggio alternativo di 160 mg alla settimana 0 e, a partire dalla settimana 2, 80 mg ogni 4 settimane (vedere «Proprietà/effetti»).
Psoriasi a placche in bambini e adolescenti (dai 6 anni)
La dose raccomandata per l'iniezione sottocutanea in pazienti pediatrici dai 6 anni e con un peso corporeo di almeno 25 kg si basa sulle seguenti categorie di peso:
Peso dei pazienti pediatrici | Dose iniziale una tantum (settimana 0) | Dose a partire dalla settimana 4 e in seguito ogni 4 settimane |
|---|---|---|
Oltre 50 kg | 160 mg (due iniezioni da 80 mg) | 80 mg (una iniezione da 80 mg) |
Da 25 a 50 kg | 80 mg (una iniezione da 80 mg) | 40 mg |
Nei pazienti pediatrici ai quali vengono prescritti 80 mg, la dose completa può essere utilizzata direttamente dalla siringa preriempita o dalla penna preriempita.
Le indicazioni per la preparazione di una siringa per iniezione con Taltz in dosi da 40 mg da una siringa preriempita da 80 mg sono disponibili in «Altre indicazioni / Indicazioni per la manipolazione».
L'utilizzo di Taltz viene sconsigliato nei bambini con un peso corporeo inferiore ai 25 kg.
Artrite psoriasica
La dose raccomandata è di 160 mg somministrata per via sottocutanea (due iniezioni da 80 mg) alla settimana 0, seguita da una dose di 80 mg (una iniezione) ogni 4 settimane.
Per i pazienti con artrite psoriasica e concomitante psoriasi a placche di grado da moderato a severo, lo schema di dosaggio raccomandato è lo stesso della psoriasi a placche.
Spondiloartrite assiale
La dose raccomandata è di 80 mg (una iniezione) somministrata con iniezione sottocutanea ogni 4 settimane.
Per la spondiloartrite assiale, durante il trattamento con Taltz possono essere utilizzati DMARD convenzionali (ad es. sulfasalazina), corticosteroidi, FANS e/o analgesici.
Durata della terapia
In tutte le indicazioni (psoriasi a placche, artrite psoriasica, spondiloartrite assiale) deve essere presa in considerazione l'interruzione del trattamento nei pazienti che non hanno mostrato alcuna risposta dopo 16 a 20 settimane di trattamento. Alcuni pazienti con una risposta iniziale parziale possono successivamente migliorare continuando il trattamento oltre le 20 settimane.
Pazienti con disturbi della funzionalità epatica
Taltz non è stato studiato in questa popolazione di pazienti.
Pazienti con disturbi della funzionalità renale
Taltz non è stato studiato in questa popolazione di pazienti.
Pazienti anziani
Dei 4204 pazienti con psoriasi a placche esposti a Taltz negli studi clinici di sviluppo, un totale di 301 pazienti aveva un'età pari o superiore a 65 anni e 36 pazienti avevano un'età pari o superiore a 75 anni. Dei 1118 pazienti con artrite psoriasica esposti a Taltz negli studi clinici, 122 avevano un'età pari o superiore a 65 anni e 6 un'età pari o superiore a 75 anni. Non sono necessari aggiustamenti della dose (vedere «Farmacocinetica»).
Bambini e adolescenti
L'efficacia e la sicurezza di Taltz in bambini di età inferiore ai 6 anni nel trattamento della psoriasi a placche non sono ancora state stabilite. Nei bambini e negli adolescenti, l'esperienza è limitata. L'efficacia e la sicurezza in caso di utilizzo oltre un anno non sono stabilite a sufficienza.
L'efficacia e la sicurezza di Taltz in bambini e adolescenti di età inferiore ai 18 anni nel trattamento dell'artrite psoriasica e della spondiloartrite assiale non sono ancora state stabilite.
Modo di somministrazione
Prima di iniziare la terapia il medico deve accertarsi che:
Il paziente o, nel caso di bambini e adolescenti, la persona con responsabilità genitoriale abbia capito che Taltz rappresenta una terapia innovativa con limitata esperienza e rischi a lungo termine sconosciuti.
Taltz viene somministrato per via sottocutanea alternando fra parte superiore del braccio, pancia e coscia.
Evitare di effettuare le iniezioni nelle aree cutanee che presentano lesioni psoriasiche.
Dopo essere stati adeguatamente istruiti alla tecnica di iniezione sottocutanea, i pazienti adulti possono iniettarsi Taltz da soli se il medico curante lo ritiene appropriato. Nel manuale di istruzioni della siringa e della penna sono riportate istruzioni dettagliate per la somministrazione.
Psoriasi a placche in bambini e adolescenti:
Nel caso di pazienti pediatrici con un peso superiore a 50 kg, le iniezioni possono essere somministrate da persone incaricate della cura dopo una formazione nella tecnica di iniezione sottocutanea.
Nel caso di pazienti pediatrici dai 25 kg ai 50 kg, le dosi di ixekizumab da 40 mg devono essere preparate e somministrate da un operatore sanitario qualificato. Utilizzare solo la siringa preriempita di Taltz 80 mg/1 ml disponibile in commercio per preparare le dosi pediatriche prescritte da 40 mg (vedere «Altre indicazioni / Indicazioni per la manipolazione»).
Dopo la somministrazione di Taltz, bambini e adolescenti devono rimanere sotto controllo medico per un periodo di tempo adeguato.
Controindicazioni
Ipersensibilità grave al principio attivo o a una qualsiasi delle sostanze ausiliarie.
Infezioni attive gravi (ad es. tubercolosi attiva, sepsi, infezioni opportunistiche gravi).
Avvertenze e misure precauzionali
Infezioni
Il trattamento con Taltz è associato a un aumento dose-dipendente del tasso di infezioni quali infezione delle vie respiratorie superiori, candidosi orale, congiuntivite e infezioni da tigna (vedere «Effetti indesiderati»). Una terapia immunomodulatoria può determinare la riattivazione di infezioni latenti.
Taltz deve essere utilizzato con cautela in pazienti con un'infezione cronica o attiva o con una storia di infezioni recidivanti. Taltz non deve essere somministrato a pazienti che presentano un'infezione attiva, in particolare le infezioni da HIV, HBV o HCV. Se si sviluppa un'infezione di questo tipo durante il trattamento con Taltz, occorre monitorare il paziente attentamente. Ai pazienti va raccomandato di consultare un medico qualora insorgano segni o sintomi di un'infezione. La terapia con Taltz deve essere interrotta se il paziente non sta rispondendo alla terapia antinfettiva standard o se l'infezione si aggrava. Il trattamento con Taltz non deve essere ripreso fino a che l'infezione non si risolve.
Taltz non deve essere somministrato a pazienti con tubercolosi attiva. Prima di iniziare Taltz in pazienti con tubercolosi latente, considerare una terapia antitubercolotica.
Neoplasie maligne
Dai dati raccolti dagli studi fino a un anno non emerge alcun aumento di rischio di neoplasie maligne. Ulteriori studi sulla sicurezza a lungo termine non sono ancora disponibili. Essendo i pazienti psoriasici una popolazione a rischio, occorre escludere la presenza di tumori cutanei nei pazienti psoriasici prima e durante il trattamento con Taltz.
Reazioni da ipersensibilità
Sono state riportate gravi reazioni di ipersensibilità, fra cui casi di anafilassi, angioedema e orticaria. Se si verifica una reazione da ipersensibilità grave, deve essere immediatamente interrotta la somministrazione di Taltz e deve essere iniziata una terapia adeguata.
Depressione
La depressione è una comorbidità nota nei pazienti con spondilite anchilosante. Si consiglia cautela in pazienti con spondilite anchilosante a rischio di depressione.
Malattie infiammatorie intestinali fra cui la malattia di Crohn e la colite ulcerosa
Con Taltz sono stati riportati casi nuovi o casi di esacerbazioni di malattie infiammatorie intestinali (vedere «Effetti indesiderati»). Taltz non è raccomandato in pazienti con una malattia infiammatoria intestinale. Se un paziente sviluppa segni e sintomi di una malattia infiammatoria intestinale o un'esacerbazione di una malattia infiammatoria intestinale già esistente, Taltz deve essere sospeso e deve essere iniziato un trattamento medico adeguato.
Immunizzazioni
Taltz non deve essere usato con vaccini vivi. Non ci sono dati disponibili sulla risposta a vaccini vivi.
Si raccomanda di completare le vaccinazione programmate prima dell'inizio del trattamento con Taltz. Questo è da osservare particolarmente in caso di utilizzo nella popolazione pediatrica.
È opportuno rispettare un intervallo di tempo fra vaccinazioni con vaccini vivi e l'inizio del trattamento secondo le direttive in materia di vaccinazione vigenti per i principi attivi immunosoppressori.
In uno studio aperto condotto su 83 soggetti sani con randomizzazione 1:1 a un gruppo di controllo o a ixekizumab 160 mg alla settimana 0 e 80 mg alla settimana 2, è stata valutata la risposta immunitaria a due vaccini inattivati contro il tetano (Boostrix®) e pneumococchi (Pneumovax-23®) somministrati alla settimana 2. Fino alla settimana 6 dopo l'immunizzazione in questo piccolo collettivo di soggetti sani, non sono stati rilevati in linea di massima problemi di sicurezza. Tuttavia, l'estrapolazione dell'efficacia e della sicurezza di vaccinazioni inattivate sulla popolazione target è possibile solo in misura limitata. I dati sulla risposta immunitaria non sono sufficienti per potere far pensare a una risposta immunitaria adeguata a tali vaccini dopo la somministrazione di Taltz nella popolazione target.
La risposta al vaccino nei bambini e negli adolescenti non è stata esaminata.
Associazione con altri medicamenti biologici
La co-somministrazione di Taltz e altri medicamenti biologici non è stata studiata e non è consigliata.
Ulteriori indicazioni
Questo medicamento contiene meno di 1 mmol di sodio (23 mg) per dose di 80 mg, cioè è essenzialmente «senza sodio».
Interazioni
Negli studi sulla psoriasi a placche, la sicurezza di Taltz in associazione con altri agenti immunomodulatori o fototerapia e vaccinazioni con vaccini vivi non è stata valutata.
Substrati del citocromo P450
I risultati di uno studio di interazione dei medicamenti, condotto su pazienti con psoriasi da moderata a severa, hanno evidenziato che la somministrazione di ixekizumab con medicamenti metabolizzati da CYP3A4 (esaminato: midazolam), CYP2C9 (esaminato: warfarin), CYP2C19 (esaminato: omeprazolo), CYP1A2 (esaminato: caffeina) o CYP2D6 (esaminato: destrometorfano) non ha un impatto clinicamente significativo sulla farmacocinetica di questi medicamenti.
Con la somministrazione di Taltz in associazione con metotressato (MTX), e/o corticosteroidi nei pazienti con artrite psoriasica, questi medicamenti non hanno avuto alcun effetto sulla farmacocinetica di Taltz.
La clearance di ixekizumab non è stata influenzata dalla somministrazione contemporanea di corticosteroidi orali, FANS o DMARD convenzionali (ad es. sulfasalazina e metotrexato) in pazienti con spondiloartrite assiale.
Gravidanza/Allattamento
Gravidanza
La quantità di dati sull'uso di ixekizumab in donne in gravidanza è limitata. È noto che le IgG umane superano la barriera placentare e ixekizumab è una IgG. Vi è quindi la possibilità che ixekizumab venga trasmesso dalla madre al feto. Durante la gravidanza e nelle donne fertili che non utilizzano un metodo contraccettivo efficace, Taltz non deve essere somministrato salvo nel caso di assoluta necessità. Alle pazienti va raccomandato di utilizzare metodi contraccettivi efficaci per almeno 10 settimane dall'ultima assunzione di Taltz.
Gli studi su animali non indicano chiari effetti dannosi diretti o indiretti su gravidanza, sviluppo embrionale/fetale, parto o sviluppo postnatale (vedere «Dati preclinici»). Non essendo sempre prevedibile la risposta nell'uomo sulla base degli studi sulla riproduzione negli animali, Taltz deve essere utilizzato durante la gravidanza solo se il beneficio è chiaramente superiore ai possibili rischi.
Allattamento
Non è noto se ixekizumab sia escreto nel latte materno o sia assorbito per via sistemica dopo l'ingestione. Tuttavia ixekizumab è escreto a bassi livelli nel latte delle scimmie Cynomolgus. Siccome molti medicamenti, fra cui gli anticorpi, vengono escreti nel latte materno, non si possono escludere rischi per i neonati/lattanti. Considerando i possibili danni per il lattante, si consiglia di non allattare durante il trattamento con Taltz e per almeno 10 settimane dopo l'ultima dose. È necessario decidere se interrompere l'allattamento al seno o la terapia con Taltz, tenendo in considerazione il beneficio dell'allattamento al seno per il bambino e quello della terapia con Taltz per la donna.
Fertilità
L'effetto di ixekizumab sulla fertilità umana non è stato valutato. Gli studi su animali non indicano effetti dannosi diretti o indiretti sulla fertilità (vedere «Dati preclinici»).
Effetti sulla capacità di condurre veicoli e sull'impiego di macchine
L'influsso di Taltz sulla capacità di condurre veicoli e sull'impiego di macchine non è stato valutato.
Effetti indesiderati
Negli studi clinici in cieco e in aperto nella psoriasi a placche, nell’artrite psoriasica, nella spondiloartrite assiale e in altre indicazioni sono stati trattati con Taltz un totale di 8956 pazienti. Di questi, 6385 pazienti sono stati esposti a Taltz per almeno un anno, rappresentando complessivamente 19’833 pazienti adulti/anno di esposizione e 207 pazienti pediatrici/anno di esposizione (196 pazienti pediatrici).
Adulti
Nella psoriasi a placche in adulti sono stati esaminati negli studi clinici un totale di 3119 pazienti (2328 pazienti con ixekizumab).
Nell'artrite proriasica in adulti sono stati esaminati negli studi clinici un totale di 678 pazienti (454 pazienti con ixekizumab).
Nella spondiloartrite assiale radiografica e non radiografica sono stati esaminati negli studi clinici un totale di 868 pazienti (574 pazienti con ixekizumab).
Il profilo di sicurezza osservato nei pazienti pediatrici con psoriasi a placche trattati ogni 4 settimane con Taltz coincide in generale con quello degli adulti (vedere paragrafo separato «Pediatria»).
Gli effetti indesiderati riportati con maggiore frequenza sono stati le reazioni sul sito di iniezione e le infezioni delle vie respiratorie superiori (tra cui la rinofaringite e la faringite). La maggior parte delle reazioni erano di intensità da leggera a moderata.
Elenco degli effetti indesiderati in studi clinici controllati con placebo (fase III, dati integrati dalle indicazioni psoriasi a placche, artrite psoriasica e spondiloartrite assiale) e segnalazioni post-marketing
Le frequenze degli effetti indesiderati vengono indicate come segue: «molto comune» (≥1/10), «comune» (≥1/100, <1/10), «non comune» (≥1/1000, <1/100), «raro» (≥1/10'000, <1/1000), «molto raro» (<1/10'000).
Infezioni ed infestazioni
Molto comune: infezioni delle vie respiratorie superiori (13.4% con Q2W e 13.5% con Q4W).
Comune: infezione da tigna.
Non comune: influenza, rinite, candidiasi orale, congiuntivite.
Raro: candidiasi esofagea.
Patologie del sistema emolinfopoietico
Non comune: neutropenia, trombocitopenia.
Disturbi del sistema immunitario
Raro: anafilassi (angioedema, edema laringeo, orticaria).
Patologie respiratorie, toraciche e mediastiniche
Comune: dolore orofaringeo.
Patologie gastrointestinali
Comune: nausea, diarrea.
Non comune: morbo di Crohn, colite ulcerosa (vedere «Avvertenze e misure precauzionali»)
Patologie epatobiliari
Comune: enzimi epatici elevati.
Patologie della cute e del tessuto sottocutaneo
Non comune: orticaria.
Patologie sistemiche e condizioni relative alla sede di somministrazione
Molto comune: reazioni nel sito di iniezione (18.2% con Q2W e 137% con Q4W).
Descrizione di effetti indesiderati selezionati
Reazioni nel sito di iniezione
Le reazioni più frequenti osservate nel sito di iniezione sono state eritema e dolore. Queste reazioni sono state prevalentemente di gravità da lieve a moderata e non hanno portato all'interruzione di Taltz.
Infezioni
La maggior parte delle infezioni erano effetti indesiderati non gravi e di gravità da lieve a moderata quali rinofaringite e infezioni delle vie respiratorie superiori che non hanno richiesto un'interruzione del trattamento.
Nel periodo di controllo con placebo degli studi clinici di fase III nella psoriasi a placche in adulti (in totale 2328 pazienti sono stati trattati con Taltz e 791 con placebo fino a 12 settimane) le infezioni sono state riportate nel 27.2% dei pazienti trattati con Taltz rispetto al 22.9% dei pazienti trattati con placebo.
Infezioni gravi si sono verificate in 13 (0,6%) dei pazienti trattati con Taltz e in 3 (0.4%) dei pazienti trattati con placebo (vedere «Avvertenze e misure precauzionali»). Gli eventi indesiderati gravi (SAE) correlati alle infezioni riportati da più di un paziente dell'intero gruppo di ixekizumab sono stati cellulite (n = 3), appendicite (n = 2) ed erisipela (n = 2). La proporzione di pazienti che hanno interrotto la terapia a seguito di eventi indesiderati legati alle infezioni è stata simile nell'insieme del gruppo ixekizumab (8 pazienti [0.3%]) e nel gruppo placebo (2 pazienti [0.3%]).
In conformità al meccanismo d'azione è stato osservato un aumento della candidosi orale e, con una sola eccezione, tutti i casi erano di gravità leggera o moderata. Non sono stati riportati SAE né interruzioni del trattamento a seguito della candidosi.
Durante tutto il periodo di trattamento, le infezioni sono state riportate nel 52.8% dei pazienti trattati con Taltz (46.9 per 100 pazienti/anno). Infezioni gravi sono state riportate nell'1.6% dei pazienti trattati con Taltz (1.5 per 100 pazienti/anno).
Neutropenia e trombocitopenia
Negli studi sulla psoriasi a placche, il 9% dei pazienti trattati con Taltz ha sviluppato neutropenia. Tali livelli di neutropenia possono persistere, oscillare o essere transitori.
Lo 0.1% dei pazienti trattati con Taltz ha sviluppato una conta dei neutrofili di < 1000 cellule/mm³. In generale la neutropenia non ha richiesto l'interruzione di Taltz.
Il 3% dei pazienti trattati con Taltz con un valore basale normale della conta delle piastrine ha mostrato una riduzione di tale conteggio a valori compresi fra 75'000 e 150'000 cellule/mm³. La trombocitopenia può persistere, oscillare o essere transitoria.
La frequenza della neutropenia e della trombocitopenia negli studi clinici sull'artrite psoriasica è paragonabile a quella osservata negli studi sulla psoriasi a placche.
Sicurezza rispetto a etanercept
Nei 2 studi clinici controllati UNCOVER-2 e UNCOVER-3 i tassi di eventi indesiderati gravi sia per Taltz che per etanercept sono stati dell'1.9%, mentre i tassi di interruzione dovuti ad eventi indesiderati sono stati dell'1.2% per etanercept e del 2% per Taltz. Il tasso di infezioni è stato del 21.5% per etanercept e del 26% per Taltz con la maggior parte degli eventi di intensità lieve o moderata. Il tasso di infezioni gravi è stato dello 0.4% per etanercept e dello 0.5% per Taltz.
Pediatria
In complesso il profilo di sicurezza nei pazienti pediatrici con psoriasi a placche, trattati ogni 4 settimane con Taltz, coincide con il profilo di sicurezza nei pazienti adulti con psoriasi a placche, ad eccezione dei seguenti effetti indesiderati, che sono stati osservati più frequentemente rispetto agli adulti o che si sono verificati specificamente in bambini e adolescenti:
Infezioni e infestazioni
Comune: influenza, congiuntivite.
Disturbi del sistema emolinfopoietico
Comune: neutropenia
Disturbi del sistema immunitario
Comune: reazioni da ipersensibilità (broncospasmo, dermatite, rash).
Patologie della cute e del tessuto sottocutaneo
Comune: orticaria.
Disordini generali e reazioni sul sito di somministrazione
Comune: febbre
Descrizione di reazioni avverse selezionate
Neutropenia
Nello studio pediatrico, in 114 pazienti con un'età di 6-17 anni e psoriasi a placche da moderata a severa, l'8.8% dei pazienti trattati con Taltz ha sviluppato una neutropenia, che può persistere, fluttuare o manifestarsi transitoriamente.
Lo 0.9% dei pazienti trattati con Taltz ha sviluppato una conta dei neutrofili di < 1000 cellule/mm³. Nello studio pediatrico Taltz non è stato sospeso in nessun paziente a causa della neutropenia.
Reazioni da ipersensibilità
Nello studio pediatrico, nel periodo di 12 settimane controllato con placebo, nel 5.2% dei pazienti nel gruppo Taltz e nell'1.8% dei pazienti nel gruppo placebo si sono verificate reazioni da ipersensibilità. Tutte le reazioni da ipersensibilità sono state di natura non anafilattica.
Malattie infiammatorie intestinali.
Malattie infiammatorie intestinali sono state osservate con frequenza non comune nei pazienti pediatrici, tuttavia sono state più frequenti rispetto agli adulti.
Nello studio pediatrico, nel periodo di 12 settimane controllato con placebo, si è verificato un morbo di Crohn nello 0.9% dei pazienti nel gruppo Taltz e nello 0% dei pazienti nel gruppo placebo. Nel periodo di tempo complessivo dello studio clinico pediatrico (fase controllata con placebo e fase di terapia di mantenimento) si è verificato un morbo di Crohn in totale in 4 soggetti trattati con Taltz (2.0%).
Reazioni nel sito di iniezione
Nello studio pediatrico sulla psoriasi a placche, nel periodo di 12 settimane controllato con placebo, nel 12.2% dei pazienti nel gruppo Taltz e nell'1.8% dei pazienti nel gruppo placebo si sono verificate reazioni nel sito di iniezione. Le reazioni osservate più di frequente nel sito di iniezione sono state dolore ed eritema. Queste reazioni sono state prevalentemente di gravità da lieve a moderata e non hanno portato all'interruzione di Taltz.
Immunogenicità
Circa il 9-17% dei pazienti adulti con psoriasi a placche trattati con Taltz allo schema di dosaggio raccomandato ha sviluppato anticorpi contro ixekizumab, la maggior parte dei quali erano con un basso titolo e non associati a una riduzione della risposta clinica fino a 60 settimane di trattamento. Tuttavia, in circa l'1% dei pazienti trattati con Taltz è stato confermato lo sviluppo di anticorpi neutralizzanti associati a basse concentrazioni di farmaco e a una riduzione della risposta clinica.
Dei pazienti pediatrici con psoriasi a placche trattati con Taltz secondo il regime di dosaggio raccomandato fino a 12 settimane, 21 pazienti (18%) hanno sviluppato anticorpi contro il principio attivo.
In circa la metà il titolo era basso e 5 pazienti (4%) avevano anticorpi neutralizzanti confermati, associati a basse concentrazioni del medicamento. Non c'è stata alcuna correlazione con risposta clinica o effetti indesiderati.
Tra i pazienti con artrite psoriasica trattati con Taltz allo schema di dosaggio raccomandato fino a 52 settimane, l'11% ha sviluppato anticorpi contro il principio attivo, la maggior parte dei quali erano a basso titolo e l'8% ha confermato lo sviluppo di anticorpi neutralizzanti. La frequenza degli anticorpi contro il principio attivo e degli anticorpi neutralizzanti è stata, rispettivamente, del 15.4% e del 10,3% nel gruppo senza somministrazione concomitante di cDMARD rispetto al 9.0% e al 6.0%, rispettivamente, nel gruppo che ha ricevuto ixekizumab in associazione con cDMARD. Non è stata osservata un'associazione chiara tra la presenza di anticorpi neutralizzanti e l'impatto sulla concentrazione del principio attivo o sull'efficacia.
Nei pazienti con spondilite anchilosante trattati con Taltz secondo il regime di dosaggio raccomandato, fino alla settimana 16 e alla settimana 52 10 pazienti (5.2%) e 23 pazienti (11.9%), rispettivamente, hanno sviluppato anticorpi contro il principio attivo, e di questi la maggioranza presentava un titolo basso; 3 pazienti (1.5%) avevano anticorpi neutralizzanti fino alla settimana 16 e nessun nuovo paziente ha sviluppato anticorpi neutralizzanti tra la settimana 16 e la settimana 52. In alcuni pazienti positivi agli anticorpi contro il principio attivo sono stati osservati livelli minimi tendenzialmente più bassi.
Nei pazienti con spondiloartrite assiale non radiografica, trattati con Taltz secondo il regime di dosaggio raccomandato fino a 52 settimane, 5 pazienti (8.9%) hanno sviluppato anticorpi contro il principio attivo e di questi tutti presentavano un titolo basso; nessun paziente presentava anticorpi neutralizzanti.
Non è stata osservata alcuna evidente correlazione tra la presenza di anticorpi contro il principio attivo e l'impatto sull'efficacia o la sicurezza.
In tutte le indicazioni, non è stata stabilita un'associazione tra immunogenicità e gli eventi indesiderati insorti durante il trattamento.
La notifica di effetti collaterali sospetti dopo l'omologazione del medicamento è molto importante. Consente una sorveglianza continua del rapporto rischio-beneficio del medicamento. Chi esercita una professione sanitaria è invitato a segnalare qualsiasi effetto indesiderato sospetto, nuovo o serio, attraverso il portale online ElViS (Electronic Vigilance System). Maggiori informazioni sul sito www.swissmedic.ch.
Posologia eccessiva
Nell'ambito di studi clinici sono state somministrate per via sottocutanea dosi fino a 180 mg senza osservare la comparsa di tossicità limitante la dose. Sono stati riportati sovradosaggi fino a 240 mg per via sottocutanea senza alcun evento indesiderato grave. In caso di sovradosaggio si raccomanda di monitorare il paziente per riscontrare eventuali segni o sintomi di effetti indesiderati e di istituire immediatamente una terapia sintomatica adeguata.
Proprietà/Effetti
Codice ATC
L04AC13
Meccanismo d'azione
Ixekizumab è un anticorpo monoclonale ricombinante umanizzato selettivo per l'interleuchina 17A. Prodotto dalle cellule CHO (Chinese Hamster Ovary), è un anticorpo monoclonale IgG4 modificato che si lega con alta affinità (< 3 pM) e specificità alla citochina proinfiammatoria interleuchina 17A (sia IL-17A che all'eterodimero IL-17A/F). Elevate concentrazioni di IL-17A sono state messe in relazione alla patogenesi di diverse malattie autoimmuni. Nella psoriasi il ligando IL-17A riveste un ruolo importante nella proliferazione e attivazione eccessive dei cheratinociti. La neutralizzazione dell'IL-17A da parte di ixekizumab inibisce queste azioni. IL-17A gioca un ruolo chiave nella patogenesi della psoriasi a placche e dell'artrite psoriasica ed è sovraregolato nella cute con lesioni rispetto alla cute senza lesioni dei pazienti con psoriasi a placche.
Nella spondiloartrite assiale il ligando IL-17A riveste un ruolo importante a causa dell'aumento delle infiammazioni, che porta a danni ossei erosivi e neoformazioni ossee patologiche.
Ixekizumab non si lega ai ligandi IL-17B, IL-17C, IL-17D, IL-17E o IL-17F.
Ixekizumab ha una scarsa capacità di legarsi ai recettori Fcγ o alle componenti del sistema del complemento. I test in vitro di legame hanno confermato che ixekizumab non si lega ai recettori umani Fcγ I, IIa e IIIa o alla componente del complemento C1q.
Farmacodinamica
Sulla base dei dati di biopsia della cute psoriasica da uno studio di fase I, dal basale al giorno 43 è stata osservata una tendenza dose-correlata alla riduzione dello spessore epidermico, del numero di cheratinociti proliferanti, delle cellule T e delle cellule dendritiche, così come una riduzione dei marker dell'infiammazione locale. Come conseguenza diretta, il trattamento con ixekizumab riduce l'eritema, l'indurimento e la desquamazione nelle lesioni della psoriasi a placche.
Taltz ha mostrato di ridurre (entro 1 settimana di trattamento) i livelli di proteina C-reattiva.
Efficacia clinica
Psoriasi a placche
L'efficacia e la sicurezza di Taltz sono state valutate in tre studi di fase III randomizzati, in doppio cieco, controllati con placebo e condotti in pazienti adulti affetti da psoriasi a placche di grado da moderato a severo che erano candidati alla fototerapia o alla terapia sistemica (UNCOVER-1, UNCOVER-2 e UNCOVER-3). L'efficacia e la sicurezza di Taltz sono state valutate anche verso etanercept (UNCOVER-2 e UNCOVER-3). I pazienti inizialmente randomizzati a Taltz che avevano risposto con un punteggio di 0 o 1 (sPGA (0,1)) alla scala static Physician Global Assessment alla settimana 12 sono stati nuovamente assegnati per randomizzazione a placebo o Taltz per ulteriori 48 settimane (UNCOVER-1 e UNCOVER-2). I pazienti randomizzati a placebo, etanercept o Taltz che non avevano risposto, non avendo raggiunto un punteggio sPGA (0,1), hanno ricevuto Taltz fino a 48 settimane. Inoltre, l'efficacia e la sicurezza a lungo termine sono state valutate in tutti e tre gli studi fino a un totale di 5 anni nei pazienti che hanno partecipato interamente agli studi.
Dei 3866 pazienti coinvolti in questi studi controllati con placebo, il 64% aveva ricevuto una precedente terapia sistemica (biologica, sistemica convenzionale o PUVA), il 43.5% aveva ricevuto una precedente fototerapia, il 49.3% aveva ricevuto una precedente terapia sistemica convenzionale e il 26.4% aveva ricevuto una precedente terapia biologica per il trattamento della psoriasi. Di tutti i pazienti, il 14.9% aveva ricevuto almeno un agente anti-TNF alfa e l'8.7% aveva ricevuto un anti-IL-12/IL-23. Il 23.4% dei pazienti aveva una storia di artrite psoriasica al basale.
Criteri di esclusione: i criteri di esclusione essenziali a una partecipazione a UNCOVER-1, UNCOVER-2 e UNCOVER-3 erano l'utilizzo concomitante di terapie psoriasiche sistemiche o biologiche o fototerapia, qualsiasi disturbo di salute o malattia grave instabile, ad eccezione della psoriasi a placche, e qualsiasi infezione grave in corso o recentemente superata. In UNCOVER-2 e UNCOVER-3 qualsiasi precedente impiego di etanercept era un criterio di esclusione.
In tutti e tre gli studi, gli endpoint co-primari sono stati la proporzione di pazienti rispetto al placebo che ha ottenuto una risposta PASI-75 (Psoriasis Area and Severity Index; PASI 75 = miglioramento del PASI di almeno il 75%) e un valore sPGA di 0 («clear») o 1 («minimal») alla settimana 12. I pazienti di tutti i gruppi di trattamento avevano un punteggio basale mediano PASI tra 17.4 e 18.3; dal 48.3% al 51.2% dei pazienti aveva un punteggio basale sPGA «grave» o «molto grave» e un punteggio basale medio della scala numerica per il prurito itch Numeric Rating Scale (itch NRS) tra 6.3 e 7.1.
Tabella 1. Risposta clinica a 12 settimane (NRI) negli studi UNCOVER-1, -2 e -3
Placebo | Taltz 80 mg Q4W | Taltz 80 mg Q2W | Etanercept 50 mg 2 volte la settimana | |
|---|---|---|---|---|
UNCOVER 1 | ||||
Numero di pazienti (N) | 431 | 432 | 433 | NA |
sPGA «0» (clear) o «1» (minimal), n (%) | 14 | 330 | 354 | NA |
sPGA «0» (clear), n (%) | 0 | 149 | 160 | NA |
PASI 75, n (%) | 17 | 357 | 386 | NA |
PASI 90, n (%) | 2 | 279 | 307 | NA |
PASI 100, n (%) | 0 | 145 | 153 | NA |
UNCOVER 2 | ||||
Numero di pazienti (N) | 168 | 347 | 351 | 358 |
sPGA «0» (clear) o «1» (minimal), n (%) | 4 | 253 | 292 | 129 |
sPGA «0» (clear), n (%) | 1 | 112 | 147 | 21 |
PASI 75, n (%) | 4 | 269 | 315 | 149 |
PASI 90, n (%) | 1 | 207 | 248 | 67 |
PASI 100, n (%) | 1 | 107 | 142 | 19 |
UNCOVER 3 | ||||
Numero di pazienti (N) | 193 | 386 | 385 | 382 |
sPGA «0» (clear) o «1» (minimal), n (%) | 13 | 291 | 310 | 159 |
sPGA «0» (clear), n (%) | 0 | 139 | 155 | 33 |
PASI 75, n (%) | 14 | 325 | 336 | 204 |
PASI 90, n (%) | 6 | 252 | 262 | 98 |
PASI 100, n (%) | 0 | 135 | 145 | 28 |
Abbreviazioni: n = numero di pazienti in questa categoria; N = numero di pazienti nella popolazione intent-to-treat; NA = non pertinente; NRI = Non-Responder Imputation; NRS = numeric rating scale; PASI = Psoriasis Area and Severity Index; Q2W = ogni due settimane; Q4W = ogni quattro settimane; sPGA = static Physician Global Assessment.
a p < 0.001 rispetto a placebo
b p < 0.001 rispetto ad etanercept
c p < 0.01 rispetto a placebo
UNCOVER-1 ha coinvolto 1296 pazienti. I pazienti sono stati randomizzati (1:1:1) per ricevere placebo o Taltz (80 mg ogni due o quattro settimane [Q2W o Q4W] dopo una dose iniziale di 160 mg) per 12 settimane.
UNCOVER-2 ha coinvolto 1224 pazienti. I pazienti sono stati randomizzati (1:2:2:2) per ricevere placebo o Taltz (80 mg ogni due o quattro settimane [Q2W o Q4W] dopo una dose iniziale di 160 mg) o etanercept 50 mg 2 volte la settimana per 12 settimane.
UNCOVER-3 ha coinvolto 1346 pazienti. I pazienti sono stati randomizzati (1:2:2:2) per ricevere placebo o Taltz (80 mg ogni due o quattro settimane [Q2W o Q4W] dopo una dose iniziale di 160 mg) o etanercept 50 mg 2 volte la settimana per 12 settimane.
Il dosaggio 80 mg Q2W ha dimostrato in tutti gli studi a tutti gli endpoint un'efficacia superiore (vedere la tabella sopra riportata) in particolare con elevati gradi di miglioramento cutaneo (PASI 90, PASI 100, sPGA 0) e con la riduzione del prurito. Sono stati dimostrati miglioramenti significativamente maggiori del basale alla settimana 12, rispetto al placebo e etanercept, nella psoriasi ungueale (valutata secondo l'indice Nail Psoriasis Severity Index (NAPSI)), nella psoriasi del cuoio capelluto (valutata secondo l'indice Psoriasis Scalp Severity Index (PSSI)) e nella psoriasi palmoplantare (valutata secondo l'indice Psoriasis Palmoplantar Severity Index (PPASI)).
Taltz è stato associato a una efficacia precoce con una riduzione > 50% del PASI medio entro la settimana 2 (Figura 1). La proporzione di pazienti che ha raggiunto un PASI 75 è stata significativamente maggiore per Taltz rispetto a placebo e a etanercept fin dalla settimana 1. Circa il 25% dei pazienti trattati con Taltz ha ottenuto un punteggio PASI < 5 entro la settimana 2, più del 55% ha ottenuto un punteggio PASI < 5 entro la settimana 4 ed è aumentato all'85% entro la settimana 12 (rispetto al 3%, 14% e 50% dei pazienti trattati con etanercept). Miglioramenti significativi della gravità del prurito sono stati osservati alla settimana 1 nei pazienti trattati con Taltz.
Taltz ha dimostrato anche un influsso favorevole sul prurito, che era significativamente superiore al placebo.
Figura 1. Miglioramento percentuale del punteggio PASI misurato ad ogni visita dopo il basale (LOCF) nella popolazione intent-to-treat durante il periodo iniziale del trattamento (UNCOVER-2 e UNCOVER-3)

L'efficacia e la sicurezza di Taltz sono state dimostrate indipendentemente da età, sesso, origine etnica, peso corporeo, gravità al basale secondo PASI e precedente trattamento con un medicamento biologico. I tassi di risposta dopo 12 settimane nei pazienti con peso corporeo < 100 kg o ≥100 kg trattati con Taltz 80 mg Q2W sono stati, rispettivamente, PASI 75 (90.1% e 85.7%) e sPGA 0 o 1 (84.5% e 75.6%). I tassi di risposta dopo 12 settimane nei pazienti con peso corporeo < 100 kg o ≥100 kg trattati con Taltz 80 mg Q4W sono stati, rispettivamente, PASI 75 (85.6% e 74.2%) e sPGA 0 o 1 (79.0% e 67.4%). La risposta non si differenziava nei pazienti affetti da psoriasi ungueale, psoriasi nell'area del viso o psoriasi sul cuoio capelluto all'inizio dello studio. Taltz è stato efficace in pazienti con precedenti trattamenti sistemici, con o senza precedente esposizione biologica/anti-TNF, compresi i pazienti che avevano fallito il trattamento con un medicamento biologico/anti-TNF. I miglioramenti degli endpoint sPGA e PASI nei pazienti con concomitante artrite psoriasica all'inizio dello studio erano simili a quelli dell'intera popolazione con psoriasi a placche da moderata a grave. Circa il 45% dei pazienti presentavano psoriasi nell'area del viso all'inizio dello studio. Di questi pazienti l'80.4% di quelli trattati con Taltz a 12 settimane erano liberi da psoriasi nell'area del viso.
Efficacia nei pazienti che non hanno risposto alla terapia con etanercept: nei pazienti identificati come non-responder a etanercept non avendo raggiunto un punteggio sPGA (0,1) alla settimana 12 nello studio UNCOVER-2 (N = 200) e successivamente trattati con Taltz 80 mg Q4W dopo un periodo di 4 settimane di washout, il 73% e l'83.5% dei pazienti sono riusciti a raggiungere, rispettivamente, un punteggio sPGA (0,1) e un PASI 75 dopo 12 settimane di trattamento.
Mantenimento della risposta alla settimana 60 e fino a 5 anni
Per valutare il mantenimento della risposta, i pazienti originariamente randomizzati a Taltz e che avevano risposto alla terapia alla settimana 12 (cioè con un punteggio sPGA di 0,1) negli studi UNCOVER-1 e UNCOVER-2 sono stati ri-randomizzati per ulteriori 48 settimane a uno dei seguenti schemi di trattamento: placebo o Taltz (80 mg ogni quattro o dodici settimane [Q4W o Q12W]). I pazienti che alla settimana 12 non avevano raggiunto un sPGA (0,1) o che durante la fase di mantenimento avevano subito una ricaduta (sPGA ≥3) hanno successivamente ricevuto Taltz 80 mg Q4W.
Nei pazienti che avevano risposto alla terapia con un punteggio sPGA (0,1) alla settimana 12 (negli studi combinati UNCOVER-1 e UNCOVER-2) la proporzione di pazienti che hanno mantenuto questa risposta alla settimana 60 era significativamente più elevata nei pazienti trattati con Taltz 80 mg Q4W (71%) rispetto ai pazienti trattati con Taltz 80 mg Q12W (35.5%) o placebo (7%).
La Tabella 2 illustra i tassi di risposta nei pazienti ri-randomizzati per ricevere la dose di mantenimento consigliata di Taltz 80 mg ogni 4 settimane in funzione della dose randomizzata all'inizio dello studio.
Tabella 2. Mantenimento della risposta ed efficacia alla settimana 60 (risultati combinati degli studi UNCOVER-1 e UNCOVER-2; NRI)
Endpoint | 80 mg Q2W (induzione)/Placebo (mantenimento) (N=211) | 80 mg Q4W (induzione)/Placebo (mantenimento) (N=191) | 80 mg Q2W (induzione)/80 mg Q4W (mantenimento) (N=221) | 80 mg Q4W (induzione)/80 mg Q4W (mantenimento) (N=195) |
|---|---|---|---|---|
Mantenimento di un sPGA «0» (clear) o «1» (minimal) | 7.6% | 6.3% | 78.3% | 68.7% |
sPGA «0» (clear) mantenuto o raggiunto | 2.8% | 1.6% | 58.8% | 49.2% |
PASI 75 | 9.0% | 7.9% | 83.3% | 74.4% |
PASI 90 | 4.7% | 4.7% | 76.5% | 66.7% |
PASI 100 | 2.8% | 1.6% | 57.5% | 49.7% |
Abbreviazioni: N = Numero di pazienti nella popolazione analizzata; NRI = Non-Responder Imputation
Il 76.4% dei pazienti che avevano risposto con un punteggio sPGA (0,1) alla settimana 12 e che erano stati assegnati alla terapia di mantenimento con Taltz 80 mg Q4W, alla settimana 60 aveva mantenuto o raggiunto un PASI < 5. Il miglioramento della gravità del prurito è stato mantenuto fino alla settimana 60 nei pazienti trattati con Taltz, che avevano risposto con un punteggio sPGA (0,1) alla settimana 12. Quanto al mantenimento della risposta fino a 60 settimane, Taltz si è rivelato efficace nei pazienti che avevano ricevuto in precedenza un trattamento sistemico, con o senza precedente esposizione ai biologici/anti-TNF, compresi quelli che avevano fallito il trattamento con un medicamento biologico/anti-TNF.
Per i pazienti che avevano risposto con un punteggio sPGA (0,1) alla settimana 12 e che erano stati ri-randomizzati al gruppo placebo, il tempo di ricaduta (sPGA ≥3) negli studi integrati UNCOVER-1 e UNCOVER-2 è stato di 164 giorni (95% CI [143, 169]). Tra questi pazienti, il 71.5% ha ottenuto nuovamente almeno una risposta sPGA di 0, 1 entro 12 settimane da quando hanno ricominciato il trattamento con Taltz 80 mg Q4W.
I miglioramenti nella psoriasi ungueale, del cuoio capelluto e palmo-plantare sono stati mantenuti fino alla settimana 60 nei pazienti trattati con Taltz che avevano risposto alla terapia con un punteggio sPGA (0,1) alla settimana 12.
Dei 591 pazienti che hanno ricevuto Taltz Q2W durante il periodo di induzione e successivamente Q4W negli studi UNCOVER-1, UNCOVER-2 e UNCOVER-3, 427 soggetti hanno completato 5 anni di trattamento con Taltz; tra questi, 101 pazienti hanno richiesto un aumento della dose. È stato osservato che, tra i pazienti che hanno completato la valutazione alla settimana 264 (N=427), 295 pazienti (69%), 289 pazienti (68%) e 205 pazienti (48%) hanno avuto, rispettivamente, una risposta sPGA (0,1), PASI 90 e PASI 100. Il DLQI è stato valutato dopo il periodo di induzione negli studi UNCOVER-1 e UNCOVER-2, 113 pazienti (66%) hanno avuto una risposta DLQI (0,1).
Qualità di vita/esiti riportati dal paziente
Alla settimana 12 (studi 1-3) Taltz è stato associato con un miglioramento statisticamente significativo rispetto al basale dell'indice DLQI (Dermatology Life Quality Index); i miglioramenti sono rimasti inalterati per 60 settimane. Taltz era associato a miglioramenti significativamente superiori dei dolori cutanei (misurati mediante la scala analogica visiva, VAS).
Studi post-marketing di comparazione diretta
L'efficacia e la sicurezza di ixekizumab sono state esaminate a confronto con ustekinumab anche nello studio a doppio cieco RHBS (IXORA-S). Ixekizumab è risultato superiore in termini di endpoint primario (risposta PASI 90 nella settimana 12, tabella 3). In tutte e tre le categorie della risposta PASI la superiorità rispetto a ustekinumab si è manifestata rapidamente. La superiorità di ixekizumab rispetto a ustekinumab è emersa altresì nei sottogruppi stratificati per peso.
Tabella 3. Tassi di risposta PASI dallo studio comparativo ixekizumab versus ustekinumab
Settimana 12 | Settimana 52 | |||
|---|---|---|---|---|
Ixekizumab* | Ustekinumab | Ixekizumab* | Ustekinumab** | |
P(n) | 136 | 166 | 136 | 166 |
PASI 75 n (%) | 120 (88.2%) | 114 (68.7%) | 120 (88.2%) | 126 (75.9%) |
PASI 90 n (%) | 99 (72.8 %)§ | 70 (42.2%) | 104 (76.5%) | 98 (59.0%) |
PASI 100 n (%) | 49 (36.0%) | 24 (14.5%) | 71 (52.2%) | 59 (35.5%) |
* Ixekizumab 160 mg è stato somministrato come dose iniziale, seguita da 80 mg nelle settimane 2, 4, 6, 8, 10 e 12, e in seguito da 80 mg Q4W
** Dosaggio basato sul peso: i pazienti trattati con ustekinumab hanno ricevuto 45 mg o 90 mg nella settimana 0 e 4, in seguito ogni 12 settimane fino alla settimana 52 (dose in base al peso secondo il dosaggio ammesso)
§ p < 0.001 versus ustekinumab (p-value solo per l'endpoint primario)
L’efficacia e la sicurezza di ixekizumab sono anche state valutate nello studio di 24 settimane, randomizzato, in doppio cieco, a gruppi paralleli RHCR (IXORA-R) che confrontava ixekizumab con guselkumab. Ixekizumab si è dimostrato superiore già alla settimana 4 nell’ottenimento di una clearance completa della cute e nell’ottenimento dell’obiettivo primario dello studio PASI 100 alla settimana 12 [ixekizumab 41.3% vs guselkumab 24.9% (p<0.001)] e non inferiore per il punteggio PASI 100 alla settimana 24 [ixekizumab 50.0% vs guselkumab 52.3% (p=0.414)].
Efficacia nella psoriasi genitale
Uno studio randomizzato, in doppio cieco, controllato con placebo (IXORA-Q) è stato condotto in 149 pazienti adulti (24% donne) con psoriasi genitale da moderata a severa (sPGA dell'area genitale ≥3). I pazienti avevano un interessamento principale pari almeno all'1% di Body Surface Area (BSA) (60.4 % un interessamento principale ≥10% BSA) e non avevano risposto ad almeno una terapia topica precedente contro la psoriasi genitale o non l'avevano tollerata. I pazienti avevano una psoriasi a placche di grado almeno moderato (definita da un punteggio sPGA ≥3 ed erano candidati alla fototerapia e/o a una terapia sistemica) da almeno 6 mesi.
I partecipanti allo studio randomizzati a Taltz hanno ricevuto una dose iniziale di 160 mg, seguita da 80 mg ogni 2 settimane per una durata di 12 settimane. L'endpoint primario era la proporzione di pazienti che avrebbero raggiunto un sPGA nell'area genitale di «0» (clear) o «1» (minimal) (sPGA-G 0/1). Alla settimana 12, indipendentemente dall'interessamento principale all'inizio dello studio, un numero significativamente maggiore di partecipanti allo studio trattati con Taltz ha raggiunto un sPGA-G 0/1 e un sPGA 0/1 rispetto ai partecipanti allo studio sotto placebo (interessamento principale all'inizio dello studio da 1% a < 10% BSA o ≥10% BSA: sPGA-G di «0» o «1»: Taltz 71% o 75%; placebo: 0% o 13%).
Psoriasi a placche pediatrica
Uno studio randomizzato, in doppio cieco, multicentrico, controllato con placebo (IXORA-Peds) ha incluso 201 pazienti pediatrici da 6 fino a meno di 18 anni di età con psoriasi a placche da moderata a severa (definita da un valore sPGA ≥3, riguardante ≥10% della superficie corporea, e un valore PASI ≥12), che erano candidati alla fototerapia o alla terapia sistemica o non erano adeguatamente controllati da una terapia topica.
I criteri di esclusione essenziali erano simili a quelli degli studi succitati con gli adulti, in particolare infezioni attive o gravi sofferte di recente nonché concomitanti terapie sistemiche convenzionali o biologiche o fototerapia.
Durante la fase controllata con placebo e con comparatore attivo, a doppio cieco, di 12 settimane, i pazienti sono stati trattati con placebo (n=56), etanercept (n=30) o Taltz (n=115), con il seguente adattamento del dosaggio in base al peso:
Taltz:
<25 kg: 40 mg nella settimana 0, in seguito 20 mg Q4W
da 25 kg a 50 kg: 80 mg nella settimana 0, in seguito 40 mg Q4W
>50 kg: 160 mg nella settimana 0, in seguito 80 mg Q4W
Etanercept:
0.8 mg/kg, massimo 50 mg per dose ogni settimana.
La risposta al trattamento è stata valutata dopo 12 settimane di terapia e definita dalla proporzione di pazienti che avevano raggiunto l'endpoint co‑primario rappresentato da un punteggio sPGA «0» (clear) o «1» (minimal), con un miglioramento di almeno 2 punti rispetto al basale (Baseline), e dalla proporzione di pazienti che avevano raggiunto una riduzione del valore PASI almeno del 75% (PASI 75) rispetto al basale.
Altri risultati valutati nella settimana 12 comprendevano la proporzione di pazienti che avevano raggiunto PASI 90, PASI 100, sPGA «0» e un miglioramento della gravità del prurito, misurata da una riduzione di almeno 4 punti su una scala di valutazione del prurito di 11 punti (itch Numeric Rating Scale, itch NRS).
I pazienti avevano un punteggio PASI basale medio di 17, con valori tra 12 e 49. Il punteggio sPGA basale nel 49% dei pazienti era severo o molto severo. Di tutti i pazienti, il 22% aveva ricevuto una precedente fototerapia, il 32% una precedente terapia sistemica convenzionale e il 4% una terapia preliminare biologica per il trattamento della psoriasi.
I dati della risposta clinica sono riportati nella tabella 4.
Tabella 4: Risultati dell'efficacia dopo 12 settimane nei pazienti pediatrici con psoriasi a placche, NRI
Endpoint | Taltza (N=115) n (%) | Placebo (N=56) n (%) | Differenza rispetto al placebo (95% CI) |
|---|---|---|---|
sPGA «0» (clear) o «1» (minimal)b | 93 (81) | 6 (11) | 70.2 (59.3, 81.0)d |
sPGA «0» (clear) | 60 (52) | 1 (2) | 50.4 (40.6, 60.2)d |
PASI 75c | 102 (89) | 14 (25) | 63.7 (51.0, 76.4)d |
PASI 90 | 90 (78) | 3 (5) | 72.9 (63.3, 82.5)d |
PASI 100 | 57 (50) | 1 (2) | 47.8 (38.0, 57.6)d |
Itch NRS (≥4 punti di miglioramento)c | 59 (71) | 8 (20) | 51.1 (35.3, 66.9)d |
Abbreviazioni: N = Numero di pazienti nella popolazione Intent-to-treat; NRI = Non-responder Imputation.
a Nella settimana 0 i pazienti hanno ricevuto 160 mg (peso corporeo [KG] >50 kg), 80 mg (KG 25-50 kg), o 40 mg (KG < 25 kg) di Taltz, in seguito 80 mg (KG >50 kg), 40 mg (KG 25-50 kg) o 20 mg (KG < 25 kg) ogni 4 settimane, per 12 settimane.
b Endpoint co-primari.
c Itch NRS (≥4 miglioramento) in pazienti con itch NRS basale ≥4. Numero di pazienti ITT con valore itch NRS basale ≥4 come segue: Taltz n = 83; Placebo n = 40.
d p<0.001
In totale 87 pazienti pediatrici con psoriasi a placche severa (PASI ≥20 o sPGA ≥4) sono stati randomizzati a ixekizumab Q4W (38 pazienti), etanercept Q1W (30 pazienti) o placebo (19 pazienti).
Alla settimana 12 si sono osservati miglioramenti per il gruppo ixekizumab Q4W rispetto al gruppo etanercept Q1W e al gruppo placebo, misurati con PASI 75 (84.2%, 63.3%, 26.3%) e sPGA (0,1) (76.3%, 53,3% e 5.3%)
Nei più importanti endpoint secondari sono emersi miglioramenti statisticamente significativi nel gruppo ixekizumab Q4W rispetto al gruppo etanercept Q1W e al gruppo placebo, misurati con: PASI 90 (76.3%, 40.0%, 0), PASI 100 (60.5%, 16.7%, 0) e sPGA (0) (63.2%, 16.7%, 0)
I pazienti nel gruppo di trattamento con ixekizumab hanno avuto una risposta CDLQI (children Dermatology Life Quality Index)/DLQI (0,1) superiore nella settimana 12 (NRI) rispetto al placebo. La differenza tra i gruppi di trattamento è emersa già a partire dalla settimana 4.
Non sono disponibili dati dopo la sospensione di Taltz nei bambini e sul rischio di riaccutizzazione della malattia.
Artrite psoriasica
La sicurezza e l'efficacia di Taltz sono state valutate in due studi di fase III, randomizzati, in doppio cieco, controllati con placebo, condotti in 780 pazienti affetti da artrite psoriasica attiva (≥3 articolazioni gonfie e ≥3 articolazioni dolenti). I pazienti in questi studi presentavano anche una diagnosi di artrite psoriasica (secondo i criteri CASPAR, Classification Criteria for Psoriatic Arthritis) per una mediana di 5.33 anni prima dell'inclusione nello studio. I pazienti randomizzati presentavano anche lesioni cutanee da psoriasi a placche (94.0%) o una storia documentata di psoriasi a placche. All'inizio dello studio il 12.1% dei pazienti soffriva di una psoriasi a placche da moderata a grave. Oltre il 58.9% e il 22.3% dei pazienti con artrite psoriasica presentava, rispettivamente, entesite o dattilite al basale. Per entrambi gli studi l'endpoint primario è stata la risposta ACR 20 (American College of Rheumatology) alla settimana 24, seguito da un periodo di estensione a lungo termine dalla settimana 24 alla settimana 156 (3 anni).
Nello studio 1 sull'artrite psoriasica (SPIRIT-P1), i pazienti naive alla terapia biologica con artrite psoriasica attiva sono stati randomizzati ai seguenti gruppi di trattamento: iniezioni sottocutanee di placebo, adalimumab 40 mg ogni 2 settimane (gruppo di controllo attivo), Taltz 80 mg ogni 2 settimane (Q2W) o 80 mg ogni 4 settimane (Q4W). Entrambi i regimi di dosaggio di Taltz prevedevano una dose iniziale di 160 mg. L'85.3% dei pazienti in questo studio aveva ricevuto un precedente trattamento con ≥1 cDMARD. Il 53% dei pazienti assumeva MTX in concomitanza con una dose media settimanale di 15.8 mg. Il 67% dei pazienti con uso concomitante di MTX ne assumeva una dose di 15 mg o maggiore. I pazienti in tutti i gruppi di trattamento con una risposta insufficiente alla settimana 16 hanno ricevuto una terapia di salvataggio (modifica della terapia di base). I pazienti trattati con Taltz Q2W o Q4W sono rimasti alla loro dose di Taltz assegnata inizialmente. I pazienti che hanno ricevuto adalimumab o placebo sono stati ri-randomizzati 1:1 a Taltz Q2W o Q4W alla settimana 16 o 24 in base alla risposta al trattamento. 243 pazienti hanno completato il periodo di estensione di 3 anni con Taltz.
Lo studio 2 sull'artrite psoriasica (SPIRIT-P2) ha arruolato pazienti che erano stati precedentemente trattati con un medicamento anti-TNF e avevano interrotto il farmaco anti-TNF per mancanza di efficacia o intolleranza (pazienti TNF-IR). I pazienti sono stati randomizzati per i seguenti gruppi di trattamento: iniezioni sottocutanee con placebo, Taltz 80 mg ogni 2 settimane (Q2W) o 80 mg ogni 4 settimane (Q4W). Entrambi i regimi di dosaggio con Taltz prevedevano una dose iniziale di 160 mg. Il 56% e il 35% dei pazienti hanno avuto una risposta insufficiente, rispettivamente, a 1 TNF o a ≥2 TNF. Lo studio SPIRIT-P2 ha valutato 363 pazienti, dei quali il 41% assumeva MTX in concomitanza con una dose media settimanale di 16.1 mg. Il 73.2% dei pazienti con uso concomitante di MTX ne assumeva una dose di 15 mg o maggiore. I pazienti in tutti i gruppi di trattamento con una risposta insufficiente alla settimana 16 hanno ricevuto una terapia di salvataggio (modifica della terapia di base). I pazienti trattati con Taltz Q2W o Q4W sono rimasti alla loro dose di Taltz assegnata inizialmente. I pazienti che hanno ricevuto placebo sono stati ri-randomizzati 1:1 a Taltz Q2W o Q4W alla settimana 16 o 24 in base alla risposta al trattamento. 168 pazienti hanno completato il periodo di estensione di 3 anni con Taltz.
Segni e sintomi
Il trattamento con Taltz ha determinato un miglioramento significativo degli indici di attività di malattia rispetto al placebo alla settimana 24 (vedere Tabella 5).
Tabella 5. Risultati di efficacia negli studi SPIRIT-P1 e SPIRIT-P2 alla settimana 24
SPIRIT-P1 | SPIRIT-P2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Endpoint | Differenza del tasso di risposta rispetto al placebo (95% CI) | Differenza del tasso di risposta rispetto al placebo (95% CI) | ||||||||
PBO (N = 106) | Taltz Q4W (N = 107) | Taltz Q2W (N = 103) | Taltz Q4W | Taltz Q2W | PBO (N = 118) | Taltz Q4W (N = 122) | Taltz Q2W (N = 123) | Taltz Q4W | Taltz Q2W | |
Risposta ACR 20, n (%) | ||||||||||
Settimana 24 | 32 (30.2) | 62 (57.9) | 64 (62.1) | 27.8 (15.0, 40.6)* | 31.9 (19.1, 44.8) * | 23 (19.5) | 65 (53.3) | 59 (48.0) | 33.8 (22.4, 45.2) * | 28.5 (17.1, 39.8) * |
Risposta ACR 50, n (%) | ||||||||||
Settimana 24 | 16 (15.1) | 43 (40.2) | 48 (46.6) | 25.1 (13.6, 36.6) * | 31.5 (19.7, 43.3) * | 6 (5.1) | 43 (35.2) | 41 (33.3) | 30.2 (20.8, 39.5) * | 28.3 (19.0, 37.5) * |
Risposta ACR 70, n (%) | ||||||||||
Settimana 24 | 6 (5.7) | 25 (23.4) | 35 (34.0) | 17.7 (8.6, 26.8) * | 28.3 (18.2, 38.5) * | 0 | 27 (22.1) | 15 (12.2) | 22.1 (14.8, 29.5) * | 12.2 (6.4, 18.0) * |
Abbreviazioni: ACR 20/50/70 = tasso di risposta 20%/50%/70% dell'American College of Rheumatology; CI = confidence interval; Q4W = Taltz 80 mg ogni 4 settimane; Q2W = Taltz 80 mg ogni 2 settimane; N = numero di pazienti nella popolazioni analizzata; n = numero di pazienti nella categoria specifica; NRI = non-responder imputation; PBO = placebo.
Avvertenza: I pazienti che alla settimana 16 hanno ricevuto la terapia di salvataggio o che avevano interrotto il trattamento o in relazione a cui mancavano dei dati, sono stati contati come non-responder per le analisi della settimana 24.
I cDMARD assunti in concomitanza hanno incluso MTX, leflunomide e sulfasalazina.
* p<0.001 rispetto al placebo.
Nei pazienti con dattilite o entesite preesistente, il trattamento con Taltz Q4W ha determinato un miglioramento della dattilite e della entesite alla settimana 24 rispetto al placebo (p<0.01).
Nei pazienti con psoriasi a placche (≥3% BSA) e artrite psoriasica concomitanti, il trattamento con Taltz Q4W ha determinato un miglioramento delle lesioni cutanee psoriasiche, come dimostrato sulla base delle risposte PASI 75, PASI 90 e PASI 100 alla settimana 24 (p < 0.001).
Nei pazienti con psoriasi a placche da moderata a grave e artrite psoriasica, lo schema di dosaggio Taltz Q2W ha mostrato un tasso di risposta al PASI 75, PASI 90 e PASI 100 significativamente superiore rispetto al placebo (p<0.001) e un beneficio clinicamente significativo rispetto allo schema di dosaggio Q4W.
Le risposte al trattamento con Taltz erano significativamente superiori rispetto a quelle al placebo già alla settimana 1 in termini di ACR 20, alla settimana 4 in termini di ACR 50 e alla settimana 8 in termini di ACR 70 e si sono protratte fino alla settimana 24; gli effetti si sono mantenuti per 3 anni nei pazienti che sono rimasti nello studio.
Figura 2. Risposta ACR 20 nello studio SPIRIT-P1 fino alla settimana 24

Per Taltz Q2W e Q4W: bp<0.01 e cp<0.001 rispetto al placebo.
Negli studi SPIRIT-P1 e SPIRIT-P2 sono state osservate risposte ACR 20/50/70 simili nei pazienti con artrite psoriasica, indipendentemente dal fatto che assumessero o no in concomitanza cDMARDs, compresa terapia con MTX.
Negli studi SPIRIT-P1 e SPIRIT-P2 sono stati dimostrati miglioramenti in tutte le componenti del punteggio ACR, compresa la valutazione del dolore da parte del paziente.
Nello studio SPIRIT-P1 l'efficacia, valutata in base a risposta ACR 20/50/70, miglioramento dell'entesite, miglioramento della dattilite e tasso di risposta PASI 75/90/100, è stata mantenuta fino alla settimana 52.
L'efficacia e la sicurezza di Taltz sono state dimostrate indipendentemente da età, genere, origine etnica, durata della malattia, peso corporeo al basale, presenza della psoriasi all'arruolamento nello studio, PCR basale, DAS28-PCR basale, uso concomitante di corticosteroidi e precedente terapia con un medicamento biologico. Taltz è stato efficace in pazienti naive ai biologici, esposti a biologici e che non avevano risposto ai biologici.
Il numero di pazienti affetti da artrite mutilante, artrite isolata con interessamento delle articolazioni interfalangee distali (IFD) e con spondilite concomitante negli studi registrativi non era sufficiente al fine di ottenere risultati significativi in questi sottogruppi specifici.
Nello studio SPIRIT-P1, 63 pazienti hanno completato 3 anni di trattamento con ixekizumab Q4W. Tra i 107 pazienti che sono stati randomizzati a ixekizumab Q4W (analisi NRI nella popolazione ITT), è stato osservato che 54 pazienti (50%), 41 pazienti (38%), 29 pazienti (27%) e 36 pazienti (34%) hanno avuto, rispettivamente, una risposta ACR20, ACR50, ACR70 e MDA alla settimana 156.
Nello studio SPIRIT-P2, 70 pazienti hanno completato 3 anni di trattamento con ixekizumab Q4W. Tra i 122 pazienti che sono stati randomizzati a ixekizumab Q4W (analisi NRI nella popolazione ITT), è stato osservato che 56 pazienti (46%), 39 pazienti (32%), 24 pazienti (20%) e 33 pazienti (27%) hanno avuto, rispettivamente, una risposta ACR20, ACR50, ACR70 e MDA alla settimana 156.
Risposta radiografica
Nello studio SPIRIT-P1, l'inibizione della progressione del danno articolare strutturale è stata valutata radiograficamente ed è stata espressa in termini di variazione del punteggio totale Sharp modificato (mTSS) e dei suoi componenti, l'indice di erosione (ES) e l'indice di restringimento della rima articolare (JSN), alle settimane 24 e 52 rispetto al basale.
La progressione dei danni articolari strutturali (mTSS) è stata inibita da Taltz rispetto al placebo alla settimana 24.
La percentuale di pazienti senza progressione dei danni articolari strutturali valutata radiograficamente (definita come variazione dell'indice mTSS dal basale ≤0.5) dalla randomizzazione fino alla settimana 24 è stata del 94.8% per Taltz Q2W, dell'89.0% per Taltz Q4W e del 77.4% per il placebo. L'inibizione dei danni strutturali con il trattamento con Taltz è stata mantenuta fino alla settimana 52.
Funzione fisica e qualità della vita correlata allo stato di salute
Negli studi SPIRIT-P1 e SPIRIT-P2 i pazienti trattati con Taltz Q2W (p<0.001) e Q4W (p<0.001) hanno presentato un miglioramento della funzionalità fisica valutato mediante l'Health Assessment Questionnaire-Disability Index (HAQ-DI) alla settimana 24, mantenuto fino alla settimana 52 nello studio SPIRIT-P1.
I pazienti trattati con Taltz hanno mostrato miglioramenti della qualità della vita correlata allo stato di salute, misurati con l'ausilio del Physical Component Summary des Short Form‑36 Health Survey (SF‑36 PCS) Score (p < 0.001). Nel Fatigue Severity NRS è stato dimostrato anche un miglioramento statisticamente significativo della stanchezza.
Spondilite anchilosante (spondiloartrite assiale radiografica, morbo di Bechterew)
La sicurezza e l'efficacia di Taltz sono state esaminate in 657 pazienti in 2 studi randomizzati, in doppio cieco, controllati con placebo (COAST-V e COAST-W) condotti in pazienti adulti ≥18 anni con spondilite anchilosante. I pazienti avevano una malattia attiva definita secondo il Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 e, nonostante una terapia con FANS, dolori alla colonna vertebrale ≥4 su una scala di valutazione numerica. In entrambi gli studi, al basale i pazienti avevano sintomi di una spondilite anchilosante in media da 17 anni (mediana di 16 anni). Al basale circa il 32% dei pazienti riceveva una terapia cDMARD concomitante (in COAST-V e in COAST-W: metotrexato 8.5% e 13.0%, sulfasalazina 28.5% e 14.6%, idrossiclorochina 0.6% e 0).
L'endpoint primario in entrambi gli studi era la percentuale di pazienti che, alla settimana 16, ha ottenuto una risposta Assessment of Spondyloarthritis International Society 40 (ASAS40).
COAST-V ha valutato 341 pazienti naïve alla terapia biologica, trattati con Taltz 80 mg o 160 mg alla settimana 0, seguiti da 80 mg ogni 2 settimane (Q2W) o ogni 4 settimane (Q4W), adalimumab 40 mg ogni 2 settimane o placebo. I pazienti trattati con placebo sono stati nuovamente randomizzati alla settimana 16 a ricevere Taltz (160 mg come dose iniziale, seguiti da 80 mg Q2W o Q4W). I pazienti trattati con adalimumab sono stati nuovamente randomizzati alla settimana 16 a ricevere Taltz (80 mg Q2W o Q4W).
COAST-W ha valutato 316 pazienti che avevano avuto esperienze precedenti con 1 o 2 inibitori del TNF (il 90% aveva una risposta insufficiente e il 10% era intollerante agli inibitori del TNF). Tutti i pazienti sono stati trattati con Taltz 80 o 160 mg alla settimana 0 e in seguito con 80 mg Q2W o Q4W o con placebo. I pazienti trattati con placebo sono stati nuovamente randomizzati alla settimana 16 a ricevere Taltz (160 mg come dose iniziale, seguiti da 80 mg Q2W o Q4W).
Risposta clinica
In entrambi gli studi, i pazienti trattati con Taltz 80 mg Q2W o 80 mg Q4W hanno dimostrato miglioramenti maggiori alla settimana 16 in termini di risposta ASAS40 e ASAS20 rispetto al placebo (tabella 6). La risposta nei pazienti era paragonabile indipendentemente dalle terapie concomitanti. In COAST-W la risposta è risultata indipendente dal numero di terapie precedenti con inibitori del TNF.
Tabella 6. Risultati dell'efficacia in COAST-V e COAST-W alla settimana 16
COAST-V, naïve alla terapia biologica | COAST-W, trattati con inibitori del TNF | ||||||
|---|---|---|---|---|---|---|---|
Taltz 80 mg Q4Wa (N=81) | Placebo (n=87) | Differenza rispetto al placebo (95% CI) | Adalimumab 40 mg Q2W (N = 90) | Taltz 80 mg Q4Wc (N=114) | Placebo (n=104) | Differenza rispetto al placebo (95% CI) | |
Risposta ASAS20b, n (%), NRI | 52 | 35 | 24.0 | 53 | 55 | 31 | 18.4 |
Risposta ASAS40b,c, n (%), NRI | 39 | 16 | 29.8 | 32 | 29 | 13 | 12.9 |
ASDAS Baseline [CFB] | 3.7 | 3.9 | -1.0 | 3.7 | 4.2 | 4.1 | -1.1 |
BASDAI Score Baseline [CFB] | 6.8 | 6.8 | -1.5 | 6.7 | 7.5 | 7.3 | -1.2 |
MRI Spine SPARCCd Baseline [CFB] | 14.5 | 15.8 | -9.5 | 20.0 | 8.3 | 6.4 | -6.3 |
BASDAI50e (%), NRI | 42 | 17 | 25 | 32* | 22 | 10 | 12 |
ASDAS <2.1 (%) (ridotta attività della malattia), NRI | 43 | 13 | 31 | 38*** | 18 | 5 | 13 |
ASAS HI Baseline [CFB] | 7.5 | 8.1 | -1.1 | 8.2 | 10.0 | 9.0 | -1.0 |
SF-36 PCS Baseline [CFB] | 34.0 [7.7] | 32.0 [3.6] | 4.1 | 33.5 [6.9]** | 27.5 [6.6]*** | 30.6 [1.4] | 5.2 |
Abbreviazioni: N = Numero di pazienti nella popolazione Intent-to-treat; NRI = Non-responder Imputation; i pazienti con dati mancati sono stati valutati come Non- Responder.
ASAS HI = Assessment of SpondyloArthritis International Society Health Index; ASDAS = Ankylosing Spondylitis Disease Activity Score; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; CFB = least square mean change from baseline nella settimana 16; MRI Spine SPARCC = Spondyloarthritis Research Consortium of Canada Magnetic Resonance Imaging Scoring of the Spine (23 discovertebral unit scale), SF-36 PCS = Short Form health survey physical component summary.
a Alla settimana 0 i pazienti hanno ricevuto 80 mg o 160 mg di Taltz.
b Risposta ASAS20: miglioramento ≥20% e miglioramento assoluto ≥1 unità (scala di 0–10) rispetto al basale in ≥3 di 4 aree (Patient Global, Spinal Pain, Function, Inflammation), senza registrare alcun peggioramento dell'area rimanente (definito come peggioramento ≥20% e peggioramento medio ≥1 unità, scala di 0–10). Risposta ASAS40: miglioramento ≥40% e miglioramento assoluto rispetto al basale ≥2 unità in ≥3 di 4 aree senza peggioramento nell'area rimanente.
c Endpoint primario.
d Numero dei pazienti ITT con dati MRT all'inizio dello studio: COAST-V: Taltz, n = 81; PBO, n = 82, ADA, n=85. COAST-W: Taltz, n = 58; PBO, n = 51.
e La riposta BASDAI50 è definita come un miglioramento ≥50% del BASDAI Score rispetto al basale.
* p<0.05; ** p<0.01; *** p<0.001 rispetto al placebo.
Il miglioramento delle componenti principali dei criteri di risposta ASAS40 (dolore alla colonna vertebrale, BASFI, valutazione globale del paziente, rigidità) e di altre misure dell'attività della malattia, incluso PCR, ha dimostrato un beneficio clinico significativo alla settimana 16.
La percentuale di pazienti che negli esami in COAST-V e COAST-W ha raggiunto una risposta ASAS40 è rappresentata nella figura 3.
Figura 3. Risposta ASAS40 in COAST-V e COAST-W fino alla settimana 16, NRIa

a I pazienti con dati mancati sono stati contati come Non-Responder.
* p<0.05; ** p<0.01; *** p<0.001 rispetto al placebo.
Nei pazienti sono stati riscontrati tassi di risposta ASAS40 paragonabili indipendentemente dai livelli di PCR all'inizio dello studio, dai punteggi ASDAS all'inizio dello studio e dai punteggi MRI Spine SPARCC. La risposta ASAS40 è stata dimostrata indipendentemente da età, genere, origine etnica, durata della malattia, peso corporeo all'inizio dello studio, punteggio BASDAI all'inizio dello studio e precedente trattamento biologico.
In COAST-V e COAST-W l'efficacia, valutata tramite gli endpoint rappresentati sopra nella settimana 16, incluse risposte ASAS20, ASAS40, ASDAS, BASDAI, BASFI e ASAS HI, è stata mantenuta fino alla settimana 52.
Risultati correlati alla salute
Già dalla settimana 1 si sono riscontrati miglioramenti nei dolori alla colonna vertebrale rispetto al placebo che si sono mantenuti fino alla settimana 16 [Taltz vs. placebo: COAST-V -3.2 vs. -1.7; COAST-W -2.4 vs. -1.0]; per quanto concerne stanchezza e mobilità della colonna vertebrale alla settimana 16 si sono registrati miglioramenti rispetto al placebo. I miglioramenti di dolori alla colonna vertebrale, stanchezza e mobilità della colonna vertebrale si sono mantenuti fino alla settimana 52.
Spondiloartrite assiale non radiografica
L’efficacia e la sicurezza di Taltz sono state valutate in uno studio randomizzato a doppio cieco con un periodo controllato con placebo di 52 settimane (COAST-X) in 303 pazienti di età ≥18 anni con spondiloartrite assiale attiva da almeno 3 mesi. I pazienti dovevano presentare segni oggettivi di infiammazione, come indicato da valori elevati della proteina C-reattiva (CRP) e/o sacroileite nella tomografia a risonanza magnetica (MRT), e nessuna indicazione radiografica definitiva di danni strutturali alle articolazioni sacroiliache. I pazienti avevano una malattia attiva come definita dal Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4, nonché dolori alla colonna vertebrale ≥4 su una Numerical Rating Scale (NRS) da 0 a 10, nonostante una terapia con FANS. I pazienti sono stati trattati con Taltz 80 mg o 160 mg nella settimana 0, seguiti da 80 mg ogni 2 settimane (Q2W) o 80 mg ogni 4 settimane (Q4W) o con placebo. L’inizio e/o l’aggiustamento della dose di medicamenti concomitanti (FANS, cDMARD, corticosteroidi, analgesici) erano ammessi dalla settimana 16.
All’inizio dello studio, i pazienti avevano sintomi di spondiloartrite assiale non radiografica in media da 11 anni. Circa il 39% dei pazienti riceveva un trattamento concomitante di cDMARD.
L’endpoint primario era la percentuale di pazienti che raggiungevano una risposta ASAS40 nella settimana 16.
Risposta clinica
Nella settimana 16, una proporzione maggiore di pazienti trattati con Taltz 80 mg Q4W ha raggiunto una risposta ASAS40 rispetto a quelli che ricevevano il placebo (tabella 7). Le risposte erano simili indipendentemente dalle terapie concomitanti.
Tabella 7. Risultati di efficacia nella settimana 16 in COAST-X, NRI
Taltz 80 mg Q4Wa (N=96) | Placebo (N=105) | Differenza rispetto al placebo (95% CI)f | |
|---|---|---|---|
Risposta ASAS20b, n (%), NRI | 52 | 41 | 15.1 |
Risposta ASAS40b,c, n (%), NRI | 34 | 20 | 16.4 |
ASDAS basale [CFB] | 3.8 | 3.8 | -0.5 |
BASDAI Score basale [CFB] | 7.0 | 7.2 | -0.7 |
MRI SIJ SPARCCd basale [CFB] | 5.1 | 6.3 | -3.1 |
ASDAS <2.1, n (%) | 26 | 13 | 15.3 |
SF-36 PCS basale [CFB] | 33.5 | 32.6 | 2.9 |
Abbreviazioni: N = Numero di pazienti nella popolazione Intent-to-treat; NRI = Non-responder Imputation, ASDAS = Ankylosing Spondylitis Disease Activity Score; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; CFB (change from baseline) = Variazioni LSM (least square mean) rispetto al basale nella settimana 16; MRI SIJ SPARCC = Spondyloarthritis Research Consortium of Canada Magnetic Resonance Imaging Scoring of the sacroiliac joint, SF-36 PCS = Short Form health survey physical component summary.
I pazienti con dati mancati sono stati valutati come Non Responder.
a Nella settimana 0 i pazienti hanno ricevuto 80 mg o 160 mg di Taltz
b Risposta ASAS20: miglioramento ≥20% e miglioramento assoluto ≥1 unità (scala di 0–10) rispetto al basale in ≥3 delle 4 aree seguenti (Patient Global, Spinal Pain, Function, Inflammation), senza registrare alcun peggioramento ≥20% e ≥1 unità (scala di 0–10) nelle aree rimanenti. Risposta ASAS40: miglioramento ≥40% e miglioramento assoluto rispetto al basale ≥2 unità in ≥3 di 4 aree senza peggioramento nell'area rimanente.
c Endpoint primario nella settimana 16.
d Numero di pazienti ITT con dati MRT all'inizio dello studio e nella settimana 16: Taltz, n = 85; PBO, n = 90.
e I pazienti con dati mancati sono stati valutati come Non Responder. Le percentuali si basano sul numero di pazienti nella popolazione ITT con ASDAS ≥2.1 all'inizio dello studio.
f I valori riportati vengono indicati come differenza in % (95% CI) per variabili categoriali e differenza in LSM (95% CI) per variabili continue.
* p<0.05; ** p<0.01; *** p<0.001 rispetto al placebo.
Il miglioramento nei principali componenti dei criteri di risposta ASAS40 (dolore alla colonna vertebrale, BASFI, valutazione globale del paziente, rigidità) e in altre misure dell’attività della malattia hanno dimostrato un significativo miglioramento clinico alla settimana 16.
La percentuale di pazienti che negli esami hanno raggiunto una risposta ASAS40 è rappresentata nella figura 4.
Figura 4. Risposta ASAS40 fino alla settimana 16 in COAST-X, NRIa
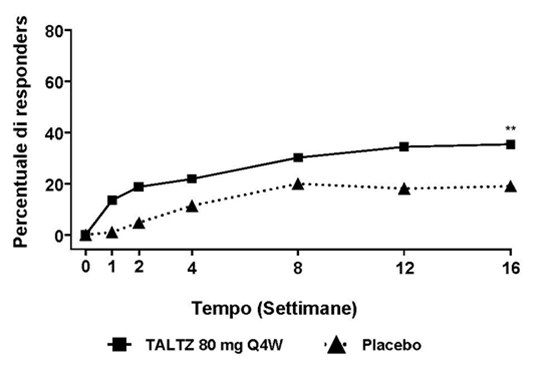
a I pazienti con dati mancati sono stati valutati come Non Responder.
** p<0.01 rispetto al placebo.
In COAST-X l'efficacia è stata mantenuta fino alla settimana 52.
Farmacocinetica
Le proprietà farmacocinetiche di Taltz nelle indicazioni psoriasi a placche, artrite psoriasica e spondiloartrite assiale sono stati paragonabili.
Assorbimento
Dopo una singola dose per via sottocutanea (SC) di ixekizumab nei pazienti con psoriasi, le concentrazioni medie massime sono state raggiunte entro 4-7 giorni, all'interno di un intervallo di dose fra 5 e 160 mg. La massima concentrazione plasmatica (Cmax) media (deviazione standard) di ixekizumab dopo la dose iniziale di 160 mg è stata 19.9 (8.15) µg/ml.
Dopo la dose iniziale di 160 mg, lo stato stazionario è stato raggiunto entro la settimana 8 con un dosaggio di 80 mg Q2W. Le Cmax,ss, e C trough,ss medie (deviazione standard) stimate sono 21.5 (9.16) µg/ml e 5.23 (3.19) µg/ml.
Dopo il passaggio dalla dose di 80 mg Q2W alla dose di 80 mg Q4W alla settimana 12, lo stato stazionario sarebbe raggiunto dopo circa 10 settimane. Le Cmax,ss, e C trough,ss medie (deviazione standard) stimate sono 14.6 (6.04) µg/ml e 1.87 (1.30) µg/ml.
Secondo le analisi effettuate, la biodisponibilità media assoluta di ixekizumab era compresa tra il 54% e il 90%. Dopo iniezione sottocutanea nell'addome (zona della pancia), nella parte superiore del braccio o nella coscia, sono state raggiunte analoghe concentrazioni sieriche di ixekizumab.
Distribuzione
Sulla base di analisi farmacocinetiche di popolazione, il volume di distribuzione totale medio allo stato stazionario è stato di 7.11 litri.
Metabolismo
Ixekizumab è un anticorpo monoclonale, per cui ci si aspetta che sia degradato in piccoli peptidi e aminoacidi attraverso le vie cataboliche nello stesso modo delle immunoglobuline G (IgG) endogene.
Eliminazione
Nell'analisi farmacocinetica di popolazione, la clearance sierica media è stata 0.0161 L/ora. La clearance è indipendente dalla dose. L'emivita media di eliminazione, come stimata dall'analisi farmacocinetica di popolazione, è di 13 giorni nei pazienti con psoriasi a placche.
Linearità/non-linearità
L'esposizione (AUC) è aumentata proporzionalmente alla dose somministrata in un range di dosaggio da 5 a 160 mg per iniezione sottocutanea.
Cinetica di gruppi di pazienti speciali
Disturbi delle funzionalità epatica
Non sono stati effettuati studi specifici di farmacologia clinica per valutare gli effetti della compromissione epatica sulla farmacocinetica di ixekizumab.
Disfunzioni renali
Non sono stati effettuati studi specifici di farmacologia clinica per valutare gli effetti della compromissione renale sulla farmacocinetica di ixekizumab.
Pazienti anziani
Sulla base dell'analisi farmacocinetica di popolazione con un numero limitato di pazienti anziani (n = 94 con un'età ≥65 anni e n = 12 con un'età ≥75 anni), la clearance nei pazienti anziani e nei pazienti con meno di 65 anni è stata simile.
Bambini e adolescenti
I pazienti pediatrici con psoriasi (età da 6 fino a meno di 18 anni) sono stati trattati per 12 settimane con ixekizumab secondo il regime di dosaggio pediatrico consigliato. Alla settimana 12, i pazienti con un peso >50 kg e da 25 a 50 kg avevano una concentrazione al livello minimo allo stato stazionario ±SD media di 3.8 ± 2.2 µg/ml o 3.9 ± 2.4 µg/ml. Queste concentrazioni erano paragonabili alle concentrazioni nei pazienti adulti con dosaggi di ixekizumab di 80 mg Q4W, con concentrazione al livello minimo allo stato stazionario ±SD media di 3.5 ± 2.2 μg/ml.
Dati preclinici
I dati non clinici ottenuti su scimmie cynomolgus non rivelano rischi particolari per l'uomo sulla base di studi di tossicità a dosi ripetute, valutazioni di sicurezza farmacologica e studi di tossicità per la riproduzione e lo sviluppo.
Tossicità per somministrazione ripetuta
La somministrazione di ixekizumab a scimmie cynomolgus per 39 settimane per via sottocutanea a dosi fino a 50 mg/kg/settimana non ha prodotto tossicità d'organo o effetti indesiderati sulla funzione immune (ad es. risposta immunitaria dipendente dalle cellule T e attività cellulare NK). La somministrazione settimanale per via sottocutanea di 50 mg/kg alle scimmie è di circa 19 volte la dose iniziale di 160 mg di Taltz e nelle scimmie determina un'esposizione (AUC) che è almeno 61 volte maggiore rispetto all'esposizione media prevista allo stato stazionario nell'uomo a cui è stato somministrato lo schema di dosaggio raccomandato.
Mutagenicità / Carcinogenicità
Non sono stati condotti studi non clinici per valutare il potenziale carcinogeno o mutageno di ixekizumab.
Tossicità per la riproduzione
Non sono stati osservati effetti sugli organi riproduttivi, i cicli mestruali o lo sperma nelle scimmie cynomolgus sessualmente mature che sono state trattate con ixekizumab per 13 settimane con una dose settimanale di 50 mg/kg per via sottocutanea.
Negli studi di tossicità per lo sviluppo, la somministrazione sottocutanea di ixekizumab a dosi fino a 50 mg/kg una volta a settimana nelle scimmie cynomolgus dall'inizio dell'organogenesi fino a poco prima della fine della gravidanza o fino alla nascita non ha indotto effetti embriotossici o teratogeni e non ha avuto effetti sul decorso del parto, né sullo sviluppo morfologico, funzionale o immunologico della prole dalla nascita fino a un'età di 6 mesi. Nelle scimmie, l'incidenza più elevata di mortalità postnatale era principalmente correlata al parto anticipato o all'incuria materna. Si tratta di risultati che sono osservati frequentemente nelle scimmie e non sono probabilmente causati da ixekizumab. Dati i numeri esigui di animali, il valore predittivo dello studio è limitato. È stato dimostrato che ixekizumab attraversa la placenta ed era presente nel sangue della prole fino all'età di 6 mesi.
Altre indicazioni
Incompatibilità
Non pertinente (medicamento in dose singola per impiego sottocutaneo).
Stabilità
Il medicamento non deve essere utilizzato oltre la data indicata con «EXP» sul contenitore
Indicazioni particolari concernenti la conservazione
Conservare in frigorifero (2-8°C). Conservare nella confezione originale per proteggere il contenuto dalla luce.
Non congelare. Taltz non deve essere utilizzato se è stato congelato.
Conservazione temporanea: Taltz può essere conservato non refrigerato fino a 5 giorni a una temperatura inferiore a 30°C.
Tenere fuori dalla portata dei bambini.
Indicazioni per la manipolazione
Non agitare.
Taltz è una soluzione sterile, senza conservanti, limpida, da incolore a leggermente gialla. Non utilizzare se si osservano particelle o se la soluzione appare torbida e/o chiaramente marrone.
La siringa preriempita e la penna preriempita sono monouso.
Le istruzioni allegate all’informazione destinata ai pazienti per l’uso della siringa o della penna preriempita devono essere seguite attentamente.
Preparazione di Taltz 40 mg per pazienti pediatrici da 25 kg a 50 kg
Le dosi di ixekizumab da 40 mg devono essere preparate e somministrate da un operatore sanitario qualificato. Utilizzare solo la siringa preriempita di Taltz 80 mg/1 ml disponibile in commercio per preparare la dose pediatrica prescritta da 40 mg.
- Iniettare l’intero contenuto della siringa preriempita in un flaconcino sterile in vetro trasparente. NON agitare o ruotare il flaconcino.
- Utilizzare una siringa monouso da 0.5 ml o 1 ml e un ago sterile per prelevare 0.5 ml dal flaconcino.
- Cambiare l’ago e utilizzare un ago sterile calibro 27 per somministrare l’iniezione al paziente. Eliminare ixekizumab non utilizzato nel flaconcino.
Ixekizumab preparato deve essere somministrato entro 4 ore dalla foratura del flaconcino sterile a temperatura ambiente.
Numero dell'omologazione
65906, 65907 (Swissmedic)
Titolare dell’omologazione
Eli Lilly (Suisse) SA, 1214 Vernier/GE.
Stato dell'informazione
Giugno 2021
Composition
Principes actifs
Ixekizumab (produit à partir de cellules CHO (Chinese Hamster Ovary) génétiquement modifiées).
Excipients
Citrate de sodium dihydraté, acide citrique, chlorure de sodium, polysorbate 80, eau pour préparation injectable q.s. ad solutionem pro 1 ml.
Quantité totale de sodium: 5.8 mg/ml.
Forme pharmaceutique et quantité de principe actif par unité
Taltz 80 mg solution injectable en seringue préremplie
Chaque seringue préremplie contient 80 mg d'ixekizumab dans 1 ml (80 mg/ml).
Taltz 80 mg solution injectable en stylo prérempli
Chaque stylo prérempli contient 80 mg d'ixekizumab dans 1 ml (80 mg/ml).
Indications/Possibilités d’emploi
Psoriasis en plaques
Taltz est indiqué dans le traitement du psoriasis en plaques modéré à sévère chez les patients adultes qui n'ont pas répondu à d'autres traitements systémiques (y compris la ciclosporine ou le méthotrexate, ou la puvathérapie) ou qui ne peuvent pas les suivre en raison de contre-indications ou d'intolérance.
Psoriasis en plaques chez les enfants et les adolescents
Taltz est indiqué dans le traitement du psoriasis en plaques modéré à sévère chez les enfants et les adolescents dès 6 ans ayant un poids corporel d'au moins 25 kg, qui n'ont pas répondu à d'autres traitements systémiques (y compris la ciclosporine ou le méthotrexate ou la puvathérapie) ou qui ne peuvent pas les suivre en raison de contre-indications ou d'intolérance.
Arthrite psoriasique
Taltz, seul ou en association avec des antirhumatismaux modificateurs de la maladie (DMARDs, disease-modifying anti-rheumatic drugs) conventionnels, est indiqué chez les patients adultes atteints d'arthrite psoriasique active n'ayant pas suffisamment répondu à un traitement par un ou plusieurs DMARDs ou ne l'ayant pas toléré.
Spondylarthrite ankylosante (maladie de Bechterew)
Taltz est indiqué dans le traitement de la spondylarthrite ankylosante active sévère chez les patients adultes n'ayant pas suffisamment répondu à une thérapie conventionnelle (par ex. anti-inflammatoires non stéroïdiens [AINS]) ou ne la tolérant pas.
Posologie/Mode d’emploi
Taltz doit être utilisé sous la conduite et la surveillance d'un médecin expérimenté dans le diagnostic et le traitement des maladies pour lesquelles Taltz est indiqué.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Posologie usuelle
Psoriasis en plaques chez les adultes
La dose recommandée est de 160 mg en injection sous-cutanée (deux injections de 80 mg) à la semaine 0, suivie de 80 mg (une injection) aux semaines 2, 4, 6, 8, 10 et 12, puis 80 mg (une injection) toutes les 4 semaines.
Chez les patients <100 kg, un schéma posologique alternatif avec 160 mg à la semaine 0 et dès la semaine 2, 80 mg toutes les 4 semaines peut être pris en considération (voir «Propriétés/Effets»).
Psoriasis en plaques chez les enfants et les adolescents (dès 6 ans)
La dose recommandée pour l'injection sous-cutanée chez les patients pédiatriques dès 6 ans ayant un poids corporel d'au moins 25 kg se base sur les catégories de poids suivantes:
Poids du patient pédiatrique | Dose initiale unique (semaine 0) | Dose dès la semaine 4 et toutes les 4 semaines par la suite |
|---|---|---|
Au-dessus de 50 kg | 160 mg (deux injections à 80 mg) | 80 mg (une injection à 80 mg) |
De 25 à 50 kg | 80 mg (une injection à 80 mg) | 40 mg |
Chez les patients pédiatriques auxquels 80 mg sont prescrits, la dose complète peut être utilisée directement à partir de la seringue préremplie ou du stylo prérempli.
Les instructions pour la préparation d'une seringue pour injecter des doses de 40 mg de Taltz à partir d'une seringue préremplie de 80 mg se trouvent sous «Remarques particulières / Remarques concernant la manipulation».
L'utilisation de Taltz n'est pas recommandée chez les enfants pesant moins de 25 kg.
Arthrite psoriasique
La dose recommandée est de 160 mg en injection sous-cutanée (deux injections de 80 mg) à la semaine 0, suivie de 80 mg (une injection) toutes les 4 semaines.
Chez les patients atteints d'arthrite psoriasique avec un psoriasis en plaques modéré à sévère concomitant, la posologie recommandée est identique à celle du traitement du psoriasis en plaques.
Spondylarthrite ankylosante
La dose recommandée est de 80 mg (une injection) en injection sous-cutanée toutes les 4 semaines.
Dans la spondylarthrite ankylosante, les DMARDs conventionnels (par ex. la sulfasalazine), les corticostéroïdes, les AINS et/ou les analgésiques peuvent être utilisés pendant le traitement par Taltz.
Durée du traitement
Dans toutes les indications (psoriasis en plaques, arthrite psoriasique, spondylarthrite ankylosante), un arrêt du traitement devrait être envisagé chez les patients qui n'ont pas répondu au bout de 16 à 20 semaines. Certains patients ayant montré une réponse initiale partielle peuvent obtenir une amélioration en poursuivant le traitement au-delà de 20 semaines.
Patients présentant des troubles de la fonction hépatique
Taltz n'a pas été étudié chez ce groupe de patients.
Patients présentant des troubles de la fonction rénale
Taltz n'a pas été étudié chez ce groupe de patients.
Patients âgés
Sur les 4204 patients atteints de psoriasis en plaques exposés à Taltz dans les études cliniques, 301 étaient âgés de 65 ans et plus, et 36 de 75 ans et plus. Sur les 1118 patients atteints d'arthrite psoriasique exposés à Taltz dans les études cliniques, 122 étaient âgés de 65 ans et plus, et 6 de 75 ans et plus. Aucune adaptation de dose n'est nécessaire (voir «Pharmacocinétique»).
Enfants et adolescents
La sécurité et l'efficacité de Taltz n'ont pas encore été établies dans le traitement du psoriasis en plaques chez les enfants de moins de 6 ans. L'expérience chez les enfants et adolescents est limitée. L'efficacité et la sécurité lors de l'utilisation au-delà d'une année n'ont pas été suffisament établies.
La sécurité et l'efficacité de Taltz n'ont pas encore été établies chez les enfants et les adolescents de moins de 18 ans dans le traitement de l'arthrite psoriasique et de la spondylarthrite ankylosante.
Mode d'administration
Avant le début du traitement, le médecin doit vérifier ce qui suit:
Le patient ou son tuteur légal pour les enfants et adolescents comprend que Taltz est un traitement inédit dont l'expérience est limitée et les risques à long terme inconnus.
Taltz est administré en injection sous-cutanée, alternativement dans le haut du bras, le ventre ou la cuisse.
Il faut éviter d'effectuer les injections dans les zones de peau présentant des lésions psoriasiques.
Après une formation adaptée à la technique d'injection sous-cutanée, les patients adultes peuvent eux-mêmes s'injecter Taltz si le médecin estime que c'est approprié. Des instructions détaillées pour l'administration sont décrites dans le manuel d'utilisation de la seringue et du stylo.
Psoriasis en plaques chez les enfants et les adolescents
Chez les patients pédiatriques pesant plus de 50 kg, le personnel soignant peut faire les injections après une formation à la technique d'injection sous-cutanée.
Pour les patients pédiatriques pesant entre 25 kg et 50 kg, les doses d'ixekizumab de 40 mg doivent être préparées et administrées par un professionnel de santé qualifié. Utilisez uniquement la seringue préremplie Taltz 80 mg/1 ml disponible dans le commerce lorsque vous préparez les doses pédiatriques prescrites de 40 mg (voir «Remarques particulières / Remarques concernant la manipulation»).
Après l'administration de Taltz, les enfants et adolescents doivent faire l'objet d'une surveillance médicale pendant une période appropriée.
Contre-indications
Hypersensibilité sévère au principe actif ou à l'un des excipients.
Infections actives graves (par exemple tuberculose active, sepsis, infection opportuniste grave).
Mises en garde et précautions
Infections
Le traitement par Taltz est associé à une augmentation dose-dépendante du taux d'infections telles que les infections des voies respiratoires supérieures, la candidose buccale, la conjonctivite et les infections fongiques à dermatophytes (teigne) (voir «Effets indésirables»). Une réactivation d'infections latentes est possible sous traitement immunomodulateur.
Taltz doit être administré avec précaution chez les patients atteints d'une infection chronique ou active ou ayant des antécédents d'infections récidivantes. Taltz ne doit pas être administré à des patients présentant une infection active, en particulier les infections à VIH, VHB ou VHC. Si une telle infection se développe pendant le traitement avec Taltz, une étroite surveillance est nécessaire. Les patients doivent être informés de la nécessité de consulter un médecin si des signes ou des symptômes indiquant une infection apparaissent. La thérapie avec Taltz doit être arrêtée en cas de non-réponse à un traitement anti-infectieux standard ainsi qu'en cas d'infection grave. Le traitement avec Taltz ne doit reprendre que lorsque l'infection a disparu.
Taltz ne doit pas être administré à des patients atteints de tuberculose active. Chez les patients présentant une tuberculose latente, il faut envisager un traitement antituberculeux avant l'instauration du traitement par Taltz.
Maladies malignes
Les études cliniques jusqu'à un an n'ont montré aucune augmentation du risque de maladies malignes. Les résultats des études concernant la sécurité à long terme ne sont pas encore disponibles. Etant donné que les patients atteints de psoriasis sont une population à risque, les patients psoriasiques doivent être examinés, avant et pendant le traitement par Taltz pour détecter la présence de tumeurs cutanées.
Réactions d'hypersensibilité
Des réactions graves d'hypersensibilité, y compris des cas d'anaphylaxie, d'angioedème et d'urticaire ont été rapportées. En cas de réaction d'hypersensibilité grave, l'administration de Taltz doit être interrompue immédiatement et un traitement approprié instauré.
Dépression
La dépression est une comorbidité connue chez les patients atteints de spondylarthrite ankylosante. La prudence est de mise chez les patients atteints de spondylarthrite ankylosante présentant un risque de dépression.
Maladie inflammatoire de l'intestin, y compris maladie de Crohn et colite ulcéreuse
Des cas de nouvelle apparition ou d'exacerbation de maladie inflammatoire de l'intestin ont été rapportés avec Taltz (voir «Effets indésirables»). Taltz n'est pas recommandé chez les patients atteints de maladie inflammatoire de l'intestin. Si un patient développe des signes et symptômes de maladie inflammatoire de l'intestin ou d'exacerbation d'une maladie inflammatoire de l'intestin préexistante, Taltz doit être interrompu et un traitement médical approprié doit être initié.
Vaccinations
Taltz ne doit pas être utilisé avec des vaccins vivants, car il n'y a pas de données disponibles concernant la vaccination avec des vaccins vivants sous Taltz.
Il est recommandé de compléter toute vaccination prévue avant le début du traitement par Taltz. Ceci est particulièrement important lors de l'utilisation dans la population pédiatrique. Il convient de respecter un certain délai entre les vaccins vivants et le début du traitement, conformément aux directives actuelles en matière de vaccination concernant les principes actifs immunosuppresseurs.
Dans une étude ouverte chez 83 volontaires sains avec randomisation 1:1 à un groupe de contrôle ou à ixekizumab 160 mg à la semaine 0 et 80 mg à la semaine 2, la réponse immunitaire a été examinée pour deux vaccins inactivés contre le tétanos (Boostrix®) et le pneumocoque (Pneumovax-23®) administrés à la semaine 2. Jusqu'à la semaine 6 après l'immunisation aucun problème de sécurité n'a en principe été constaté dans ce petit groupe de volontaires sains. Cependant, l'extrapolation de l'efficacité et la sécurité des vaccins inactivés à la population cible n'est possible que de manière limitée. Les données sur la réponse immunitaire n'étaient pas suffisantes pour pouvoir conclure à une réponse immunitaire adéquate à ces vaccins après l'administration de Taltz dans la population cible.
La réponse vaccinale n'a pas été étudiée chez les enfants et adolescents.
Association à d'autres médicaments biologiques
L'administration concomitante de Taltz avec d'autres médicaments biologiques n'a pas été étudiée et n'est pas recommandée.
Remarque complémentaire
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose de 80 mg, c.-à-d. qu'il est essentiellement «sans sodium».
Interactions
Dans les études sur le psoriasis en plaques, la sécurité de Taltz en association avec d'autres agents immunomodulateurs ou de la photothérapie ainsi qu'avec des vaccins vivants n'a pas été étudiée.
Substrats du cytochrome P450
Les résultats d'une étude d'interaction médicamenteuse chez des patients présentant un psoriasis modéré à sévère ont montré que l'administration d'ixekizumab avec des médicaments métabolisés par le CYP3A4 (étudié: le midazolam), le CYP2C9 (étudié: la warfarine), le CYP2C19 (étudié: l'oméprazole), le CYP1A2 (étudié: la caféine) ou le CYP2D6 (étudié: le dextrométhorphane) n'a pas d'impact cliniquement significatif sur la pharmacocinétique de ces médicaments.
Lors de l'administration de Taltz en association avec le méthotrexate (MTX) et / ou des corticostéroïdes chez des patients atteints d'arthrite psoriasique, ces médicaments n'avaient aucun effet sur la pharmacocinétique de Taltz.
La clairance de l'ixekizumab n'a pas été affectée par l'administration concomitante de corticostéroïdes oraux, d'AINS ou de DMARDs conventionnels (par ex. la sulfasalazine et le méthotrexate) chez les patients atteints de spondylarthrite ankylosante.
Grossesse/Allaitement
Grossesse
Il existe des données limitées concernant l'utilisation de l'ixekizumab chez la femme enceinte. Il est connu que les IgG humaines traversent la barrière placentaire et ixekizumab est une IgG. Par conséquent, il est possible que l'ixekizumab passe de la mère au fœtus. Pendant la grossesse et chez les femmes en âge de procréer qui n'utilisent pas de contraception efficace, Taltz ne doit pas être administré, à moins d'une nécessité absolue. Les patientes doivent être informées d'utiliser des méthodes contraceptives efficaces pendant au moins 10 semaines après la dernière dose de Taltz.
Les études réalisées sur l'animal n'ont pas mis clairement en évidence d'effets délétères directs ou indirects sur la grossesse, le développement embryonnaire/fœtal, l'accouchement ou le développement post-natal (voir «Données précliniques»). Comme la réponse chez l'homme ne peut pas toujours être prédite à partir des études de reproduction chez l'animal, Taltz ne doit être utilisé pendant la grossesse que si le bénéfice l'emporte clairement sur les risques.
Allaitement
On ne sait pas si l'ixekizumab est excrété dans le lait maternel chez l'être humain ou s'il passe dans la circulation sanguine après ingestion. Cependant, l'ixekizumab est excrété à de faibles taux dans le lait des guenons Cynomolgus. Comme beaucoup de médicaments, dont les anticorps, sont excrétés dans le lait maternel, un risque pour le nouveau-né / l'enfant en bas âge ne peut pas être exclu. En raison de possible dommages pour le nourrisson, il est recommandé de ne pas allaiter pendant le traitement avec Taltz et pendant au-moins 10 semaines après la dernière dose. Il convient donc de décider soit d'arrêter l'allaitement, soit d'interrompre le traitement par Taltz en tenant compte du bénéfice de l'allaitement pour l'enfant et de celui du traitement pour la mère.
Fertilité
L'effet de l'ixekizumab sur la fertilité chez l'être humain n'a pas été évalué. Les études réalisées sur l'animal n'ont pas mis en évidence d'effets délétères directs ou indirects sur la fertilité (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machines
L'effet de Taltz sur l'aptitude à conduire des véhicules ou à utiliser des machines n'a pas été étudié.
Effets indésirables
Au total, 8956 patients ont été traités par Taltz au cours des études cliniques en aveugle et en ouvert dans le psoriasis en plaques, l'arthrite psoriasique, la spondylarthrite ankylosante et dans d'autres indications. Sur l'ensemble de ces patients, 6385 patients ont été exposés à Taltz pendant au moins un an, ce qui représente une exposition cumulée de 19'833 patients-années adultes et 207 patients-années pédiatriques (196 patients pédiatriques).
Adultes
Dans le psoriasis en plaques chez les adultes, trois études de phase III contrôlées versus placebo ont été intégrées pour évaluer la sécurité de Taltz par rapport au placebo jusqu'à 12 semaines après le début du traitement. Au total, 3119 patients ont été évalués (1161 patients à la dose de 80 mg toutes les 4 semaines (1x/4 sem.), 1167 patients à la dose de 80 mg toutes les 2 semaines (1x/2 sem.) et 791 patients sous placebo).
Dans deux de ces études de phase III la sécurité de Taltz a aussi été comparée à étanercept jusqu'à 12 semaines après le début du traitement (729 patients sous 80 mg 1x/4sem., 734 patients sous 80 mg 1x/2 sem. et 739 patients sous étanercept).
Le profil de sécurité observé chez les patients pédiatriques atteints de psoriasis en plaques traités par Taltz toutes les 4 semaines est globalement cohérent avec celui des adultes (voir paragraphe séparé sur la pédiatrie).
Dans l'arthrite psoriasique chez les adultes, deux études de phase III contrôlées versus placebo ont été intégrées pour évaluer la sécurité de Taltz par rapport au placebo jusqu'à 24 semaines après le début du traitement. Au total, 678 patients ont été évalués (229 patients à la dose de 80 mg toutes les 4 semaines (1x/4 sem.), 225 patients à la dose de 80 mg toutes les 2 semaines (1x/2 sem.) et 224 patients sous placebo). Le profil de sécurité chez les patients atteints d'arthrite psoriasique est comparable à celui des patients atteints de psoriasis en plaques.
Dans la spondylarthrite ankylosante, Taltz a été évalué dans deux études contrôlées versus placebo. Au total, 657 patients ont été évalués (376 patients avec Taltz et 191 avec placebo). Au total, 195 patients dans ces études ont reçu 80 ou 160 mg de Taltz à la semaine 0, puis 80 mg toutes les 4 semaines (1x/4 sem.). Dans l'ensemble, le profil de sécurité observé chez les patients atteints de spondylarthrite ankylosante avec Taltz 1x/4 sem. est comparable à celui des patients atteints de psoriasis en plaques.
Les effets indésirables les plus fréquemment rapportés ont été des réactions au site d'injection et des infections des voies respiratoires supérieures (y compris la nasopharyngite et la pharyngite). La plupart des réactions étaient d'intensité légère à modérée.
Liste des effets indésirables observés dans les études cliniques (phase III, données intégrées des indications psoriasis en plaques, arthrite psoriasique et spondylarthrite ankylosante) et issus d'annonces après la mise sur le marché
La fréquence des effets indésirables est indiquée comme suit: «très fréquents» (≥1/10), «fréquents» (≥1/100, <1/10), «occasionnels» (≥1/1000, <1/100), «rares» (≥1/10'000, <1/1000), «très rares» (<1/10'000).
Infections et infestations
Très fréquent: infection des voies respiratoires supérieures (13.4% avec 1x/2 sem. et 13.5% avec 1x/4 sem.).
Fréquent: infection fongique à dermatophytes (teigne).
Occasionnel: grippe, rhinite, candidose buccale, conjonctivite.
Rare: candidose oesophagienne.
Affections hématologiques et du système lymphatique
Occasionnel: neutropénie, thrombocytopénie.
Affections du système immunitaire
Rare: anaphylaxie (angioedème, oedème du larynx, urticaire).
Affections respiratoires, thoraciques et médiastinales
Fréquent: douleurs oropharyngées.
Affections gastro-intestinales
Fréquent: nausées, diarrhée.
Occasionnel: maladie de Crohn, colite ulcéreuse (voir «Mises en garde et précautions»).
Affections hépatobiliaires
Fréquent: enzymes hépatiques élevées.
Affections de la peau et du tissu sous-cutané
Occasionnel: urticaire.
Troubles généraux et anomalies au site d'administration
Très fréquent: réactions au site d'injection (18.2% avec 1x/2 sem et 13.7% avec 1x/4 sem).
Description de certains effets indésirables
Réactions au site d'injection
Les réactions au site d'injection les plus fréquemment observées étaient érythème et douleurs. Pour la majorité, ces réactions ont été d'intensité légère à modérée et n'ont pas donné lieu à l'arrêt de Taltz.
Infections
La majorité des infections étaient des effets indésirables non graves et d'intensité légère à modérée, tels que rhinopharyngite et infections des voies respiratoires supérieures qui n'ont pas nécessité l'arrêt du traitement.
Pendant la période contrôlée versus placebo des études cliniques de phase III dans le psoriasis en plaques chez l'adulte (au total 2328 patients ont été traités avec Taltz et 791 patients avec placebo jusqu'à 12 semaines), des infections ont été rapportées chez 27.2 % des patients traités par Taltz pendant une durée maximale de 12 semaines, contre 22.9 % des patients traités par placebo.
Des infections graves sont survenues chez 13 (0.6%) des patients recevant Taltz et 3 (0.4%) des patients recevant le placebo (voir «Mises en garde et précautions»). Les événements indésirables graves (SAE) liés aux infections, rapportés chez plus de 1 patient de l'ensemble du groupe ixekizumab, étaient cellulite (n = 3), appendicite (n = 2) et érysipèle (n = 2). La proportion de patients qui ont interrompu le traitement en raison d'effets indésirables liés aux infections, était similaire dans l'ensemble du groupe ixekizumab (8 patients [0.3%]) et dans le groupe placebo (2 patients [0.3%]).
Conformément au mécanisme d'action, on a observé une augmentation de candidose orale et, à une exception près, tous les cas étaient de gravité légère ou modérée. Aucun SAE ni arrêt de traitement n'a été rapporté en raison de candidose.
Sur la durée totale du traitement, les infections ont été rapportées chez 52.8 % des patients traités par Taltz (46.9 pour 100 patients-année). Les infections graves ont été rapportées chez 1.6 % des patients traités par Taltz (1.5 pour 100 patients-année).
Neutropénie et thrombocytopénie
Dans les études sur le psoriasis en plaques, 9 % des patients traités par Taltz ont développé une neutropénie. De tels niveaux de neutropénie peuvent persister, fluctuer ou être transitoires.
0.1 % des patients ayant reçu Taltz ont eu un taux de neutrophiles < 1000 cellules/mm3. En général, la neutropénie n'a pas nécessité l'arrêt de Taltz.
3 % des patients exposés à Taltz ayant une valeur initiale normale de plaquettes ont montré une diminution à des valeurs comprises entre 75'000 et 150'000 cellules/mm3. La thrombocytopénie peut persister, fluctuer ou être transitoire.
La fréquence des neutropénies et des thrombocytopénies dans les études cliniques sur l'arthrite psoriasique est similaire à celle observée dans les études sur le psoriasis en plaques.
Sécurité en comparaison à étanercept
Dans les 2 études cliniques contrôlées comprenant un comparateur actif UNCOVER-2 et UNCOVER-3, le taux d'événements indésirables graves a été de 1.9 % dans le groupe étanercept comme dans le groupe Taltz et le taux d'arrêt pour cause d'événement indésirable a été de 1.2 % dans le groupe étanercept et de 2.0 % dans le groupe Taltz. Le taux d'infections a été de 21.5 % dans le groupe étanercept et de 26.0 % dans le groupe Taltz, avec la majorité des événements qui était d'intensité légère à modérée. Le taux d'infections graves a été de 0.4 % dans le groupe étanercept et de 0.5 % dans le groupe Taltz.
Pédiatrie
Dans l'ensemble, le profil de sécurité chez les patients pédiatriques atteints de psoriasis en plaques traités par Taltz toutes les 4 semaines est cohérent avec le profil de sécurité chez les patients adultes atteints de psoriasis en plaques, à l'exception des effets indésirables suivants, qui ont été observés plus fréquemment que chez les adultes, ou sont survenus spécifiquement chez les enfants et les adolescents:
Infections et infestations
Fréquent: grippe, conjonctivite.
Affections hématologiques et du système lymphatique
Fréquent: neutropénie.
Affections du système immunitaire
Fréquent: réactions d'hypersensibilité (bronchospasme, dermatite, rash).
Affections de la peau et du tissu sous-cutané
Fréquent: urticaire.
Troubles généraux et anomalies au site d'administration
Fréquent: fièvre.
Description de certains effets indésirables
Neutropénie
Dans l'étude pédiatrique chez 114 patients entre 6 et 17 ans avec psoriasis en plaques modéré à sévère, 8.8% des patients traités par Taltz ont développé une neutropénie qui peut persister, fluctuer ou être transitoire.
0.9 % des patients traités par Taltz ont eu un taux de neutrophiles < 1000 cellules/mm3. Dans l'étude pédiatrique, aucun patient n'a arrêté le traitement avec Taltz en raison d'une neutropénie.
Réactions d'hypersensibilité
Dans l'étude pédiatrique, 5.2% des patients du groupe Taltz et 1.8% des patients du groupe placebo ont présenté des réactions d'hypersensibilité durant la période de 12 semaines contrôlée contre placebo. Toutes les réactions d'hypersensibilité étaient de nature non anaphylactique.
Maladie inflammatoire de l'intestin
Des maladies inflammatoires de l'intestin ont été observées occasionnellement chez les patients pédiatriques mais étaient toutefois plus fréquentes que chez les adultes.
Dans l'étude pédiatrique, une maladie de Crohn est survenue chez 0.9 % des patients du groupe Taltz et 0 % des patients du groupe placebo durant la période de 12 semaines contrôlée contre placebo. Une maladie de Crohn est apparue chez un total de 4 patients (2.0 %) traités par Taltz sur toute la durée de l'étude pédiatrique (phase contrôlée par placebo et de maintenance).
Réactions au site d'injection
Dans l'étude pédiatrique sur la psoriasis en plaques, des réactions au site d'injection sont survenues chez 12.2 % des patients du groupe Taltz et 1.8 % des patients du groupe placebo durant la période de 12 semaines contrôlée contre placebo. Les réactions au site d'injection les plus fréquemment observées étaient douleurs et érythème. Pour la majorité, ces réactions ont été d'intensité légère à modérée et n'ont pas donné lieu à l'arrêt de Taltz.
Immunogénicité
Environ 9 à 17 % des patients adultes atteints de psoriasis en plaques traités par Taltz à la posologie recommandée ont développé des anticorps anti-ixekizumab; dans la majorité des cas, les titres étaient faibles et n'ont pas été associés à une diminution de la réponse clinique jusqu'à 60 semaines de traitement. Cependant, environ 1 % des patients traités par Taltz ont présenté des anticorps neutralisants associés à de faibles concentrations du médicament et à une baisse de la réponse clinique.
Parmi les patients pédiatriques atteints de psoriasis en plaques traités par Taltz à la posologie recommandée jusqu'à 12 semaines, 21 patients (18%) ont développé des anticorps anti-médicament.
Chez environ la moitié d'entre eux les titres étaient faibles, et 5 patients (4%) ont présenté des anticorps neutralisants associés à de faibles concentrations du médicament. Il n'y avait pas d'association avec la réponse clinique ou les événements indésirables.
Chez les patients atteints d'arthrite psoriasique traités par Taltz à la posologie recommandée jusqu'à 52 semaines, 11 % ont développé des anticorps anti-médicament, dans la majorité des cas les titres étaient faibles et 8 % ont présenté des anticorps neutralisants. L'incidence d'anticorps contre le médicament et d'anticorps neutralisants était de 15.4 % et 10.3 %, respectivement, dans le groupe sans administration simultanée de cDMARDs contre 9.0 % et 6.0 % dans le groupe recevant l'ixékizumab en association avec les cDMARDs. Aucune corrélation claire n'a été observée entre la présence d'anticorps neutralisants et un effet sur la concentration ou l'efficacité du médicament.
Chez les patients atteints de spondylarthrite ankylosante traités par Taltz à la posologie recommandée, 10 patients (5.2 %) à la semaine 16 et 23 patients (11.9 %) à la semaine 52 ont développé des anticorps anti-médicament dont la majorité avait un faible titre; 3 patients (1.5 %) avaient des anticorps neutralisants à la semaine 16, et aucun nouveau patient n'a développé d'anticorps neutralisants entre la semaine 16 et la semaine 52. Une tendance à des taux résiduels plus faibles a été observée chez certains patients positifs aux anticorps anti-médicaments. Aucun lien évident n'a pu être observé entre la présence d'anticorps anti-médicament et une influence sur l'efficacité ou la sécurité.
Dans toutes les indications, il n'a pas été établi de lien entre l'immunogénicité et les événements indésirables apparus pendant le traitement.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
Des doses jusqu'à 180 mg ont été administrées par voie sous-cutanée dans le cadre d'essais cliniques sans apparition de toxicité dose-limitante. Des surdosages jusqu'à 240 mg par voie sous-cutanée ont été rapportés sans apparition d'événement indésirable grave. En cas de surdosage, il est recommandé de surveiller l'apparition de signes ou de symptômes de réactions indésirables et d'instaurer immédiatement un traitement symptomatique.
Propriétés/Effets
Code ATC
L04AC13
Mécanisme d'action
L'ixekizumab est un anticorps monoclonal recombinant humanisé sélectif pour l'interleukine 17-A. L'ixekizumab, produit par des cellules ovariennes de hamster chinois (CHO), est un anticorps monoclonal IgG4 modifié qui se lie spécifiquement et avec une affinité élevée (< 3 pM) à la cytokine pro-inflammatoire interleukine 17A (à la fois à l'IL‑17A et à l'hétérodimère IL‑17A/F). Des concentrations élevées d'IL-17A ont été associées à la pathogenèse de diverses maladies autoimmunes. Dans le psoriasis, le ligand IL-17A joue un rôle important dans la prolifération et l'activation excessives des kératinocytes. La neutralisation de l'IL‑17A par l'ixekizumab inhibe ces phénomènes. L'IL-17A joue un rôle clé dans la pathogenèse du psoriasis en plaques et de l'arthrite psoriasique et est régulée à la hausse dans la peau affectée par les lésions par opposition à la peau non lésionnelle des patients atteints de psoriasis en plaques.
Dans la spondylarthrite ankylosante, le ligand IL-17A joue un rôle important de par l'augmentation de l'inflammation, qui entraîne des lésions osseuses érosives et une nouvelle formation osseuse pathologique.
L'ixekizumab ne se lie pas aux ligands IL-17B, IL-17C, IL-17D, IL-17E ni IL-17F.
L'ixekizumab a une faible capacité de liaison aux récepteurs Fcγ ou aux composants du complément. Les essais de liaison in vitro ont confirmé que l'ixekizumab ne se lie pas aux récepteurs humains Fcγ I, IIa et IIIa ni au composant du complément C1q.
Pharmacodynamique
Sur la base des données des biopsies de peau psoriasique issues d'une étude de phase I, on observe une tendance dose-dépendante en faveur d'une diminution de l'épaisseur de l'épiderme, du nombre de kératinocytes, de lymphocytes T et de cellules dendritiques en prolifération, ainsi que des diminutions des marqueurs d'inflammation locale entre le stade initial et le jour 43. En conséquence, le traitement par ixekizumab réduit l'érythème, l'induration et la desquamation dans les lésions de psoriasis en plaques.
Il a été observé que Taltz réduisait (après 1 semaine de traitement) les taux de protéine C réactive.
Efficacité clinique
Psoriasis en plaques
L'efficacité et la sécurité de Taltz ont été évaluées dans le cadre de trois études de phase III randomisées, en double-aveugle, contrôlées versus placebo chez des patients adultes atteints de psoriasis en plaques modéré à sévère, candidats à la photothérapie ou à un traitement systémique (UNCOVER-1, UNCOVER-2 et UNCOVER-3). L'efficacité et la sécurité de Taltz ont également été évaluées versus étanercept (UNCOVER-2 et UNCOVER-3). Les patients randomisés dans le groupe Taltz qui, à la 12ème semaine, étaient répondeurs selon le score sPGA (static Physician Global Assessment ou Evaluation Globale du Médecin) de 0 ou de 1 (0,1) ont été à nouveau randomisés dans le groupe placebo ou Taltz pendant 48 semaines supplémentaires (UNCOVER-1 et UNCOVER-2). Les patients randomisés dans le groupe placebo, étanercept ou Taltz, non-répondeurs selon le score sPGA (0,1), ont été traités par Taltz jusqu'à 48 semaines.
Sur les 3866 patients inclus dans ces études contrôlées versus placebo, 64 % avaient reçu un traitement systémique antérieur (biologique, systémique conventionnel ou psoralène et ultraviolets A (PUVA)), 43.5 % avaient déjà été traités par photothérapie, 49.3 % avaient déjà reçu un traitement systémique conventionnel et 26.4 % avaient déjà reçu une biothérapie pour le traitement du psoriasis. Sur l'ensemble de ces patients, 14.9 % avaient reçu au moins un agent anti-TNF alfa et 8.7 % un anti-IL-12/IL-23. A l'inclusion 23.4 % des patients avaient des antécédents d'arthrite psoriasique.
Critères d'exclusion: les critères d'exclusion essentiels qui s'opposaient à la participation dans UNCOVER-1, UNCOVER-2 et UNCOVER-3 étaient l'utilisation concomitante de traitements anti-psoriasiques systémiques ou biologiques ou la photothérapie, tout trouble médical grave ou maladie instables, sauf le psoriasis en plaques, et toute infection grave actuelle ou récente. Dans UNCOVER-2 et UNCOVER-3, l'utilisation précédente d'étanercept était un critère d'exclusion.
Dans les trois études, les co-critères d'évaluation principaux étaient la proportion de patients ayant atteint une réponse PASI 75 (Psoriasis Area and Severity Index; PASI 75 = amélioration du PASI d'au-moins 75 %) et une réponse sPGA 0 («blanchi») ou 1 («minimal») à la 12ème semaine par rapport au placebo. A l'inclusion, les patients de tous les groupes de traitement présentaient un score PASI médian compris entre 17.4 et 18.3; 48.3 % à 51.2 % des patients présentaient un score sPGA initial «sévère» ou «très sévère» et un score de prurit moyen compris entre 6.3 et 7.1 sur l'échelle d'évaluation numérique des démangeaisons (itch NRS, itch Numeric Rating Scale).
Tableau 1: réponse clinique après 12 semaines (NRI) dans les études UNCOVER-1, -2 et -3
Placebo | Taltz | Taltz | Etanercept | |
|---|---|---|---|---|
UNCOVER 1 | ||||
Nombre de patients (N) | 431 | 432 | 433 | NA |
sPGA de «0» (blanchi) ou de «1» (minimal), n (%) | 14 (3.2%) | 330 (76.4%)a | 354 (81.8%)a | NA |
sPGA de «0» (blanchi), n (%) | 0 | 149 (34.5%)a | 160 (37.0%)a | NA |
PASI 75, n (%) | 17 (3.9%) | 357 (82.6%)a | 386 (89.1%)a | NA |
PASI 90, n (%) | 2 (0.5%) | 279 (64.6%)a | 307 (70.9%)a | NA |
PASI 100, n (%) | 0 | 145 (33.6%)a | 153 (35.3%)a | NA |
UNCOVER 2 | ||||
Nombre de patients (N) | 168 | 347 | 351 | 358 |
sPGA de «0» (blanchi) ou de «1» (minimal), n (%) | 4 (2.4%) | 253 (72.9%)a | 292 (83.2%)a | 129 (36.0%)a |
sPGA de «0» (blanchi), n (%) | 1 (0.6%) | 112 (32.3%)a,b | 147 (41.9%)a,b | 21 (5.9%)c |
PASI 75, n (%) | 4 (2.4%) | 269 (77.5%)a | 315 (89.7%)a | 149 (41.6%)a |
PASI 90, n (%) | 1 (0.6%) | 207 (59.7%)a,b | 248 (70.7%)a,b | 67 (18.7%)a |
PASI 100, n (%) | 1 (0.6%) | 107 (30.8%)a,b | 142 (40.5%)a,b | 19 (5.3%)c |
UNCOVER 3 | ||||
Nombre de patients (N) | 193 | 386 | 385 | 382 |
sPGA de «0» (blanchi) ou de «1» (minimal), n (%) | 13 (6.7%) | 291 (75.4%)a,b | 310 (80.5%)a,b | 159 (41.6%)a |
sPGA de «0» (blanchi), n (%) | 0 | 139 (36.0%)a,b | 155 (40.3%)a,b | 33 (8.6%)a |
PASI 75, n (%) | 14 (7.3%) | 325 (84.2%)a,b | 336 (87.3%)a,b | 204 (53.4%)a |
PASI 90, n (%) | 6 (3.1%) | 252 (65.3%)a,b | 262 (68.1%)a,b | 98 (25.7%)a |
PASI 100, n (%) | 0 | 135 (35.0%)a,b | 145 (37.7%)a,b | 28 (7.3%)a |
Abréviations: n = nombre de patients dans cette catégorie; N = nombre de patients dans la population en intention de traiter: NA = non applicable; NRI = Non-Responder Imputation; NRS = numeric rating scale; PASI = Psoriasis Area and Severity Index; 1x/2 sem. = toutes les 2 semaines; 1x/4 sem. = toutes les 4 semaines; sPGA = static Physician Global Assessment.
a p < 0.001 versus au placebo
b p < 0.001 versus étanercept
c p < 0.01 versus placebo
L'étude UNCOVER-1 incluait 1296 patients. Les patients ont été randomisés (1:1:1) pour recevoir pendant 12 semaines le placebo ou Taltz (80 mg toutes les deux ou quatre semaines [1x/2 sem. ou 1x/4 sem.] après une dose initiale de 160 mg).
L'étude UNCOVER-2 incluait 1224 patients. Les patients ont été randomisés (1:2:2:2) pour recevoir pendant 12 semaines le placebo ou Taltz (80 mg toutes les deux ou quatre semaines [1x/2 sem. ou 1x/4 sem.] après une dose initiale de 160 mg) ou de l'étanercept 50 mg deux fois par semaine.
L'étude UNCOVER-3 incluait 1346 patients. Les patients ont été randomisés (1:2:2:2) pour recevoir pendant 12 semaines le placebo ou Taltz (80 mg toutes les deux ou quatre semaines [1x/2 sem. ou 1x/4 sem.] après une dose initiale de 160 mg) ou de l'étanercept 50 mg deux fois par semaine.
Le dosage de 80 mg 1x/2 sem. a démontré dans toutes les études pour tous les critères d'évaluation une efficacité supérieure (voir tableau ci-dessus), en particulier dans les degrés élevés d'amélioration de la peau (PASI 90, PASI 100, sPGA 0) et dans la réduction des démangeaisons. En comparaison avec le placebo et l'étanercept, à la semaine 12, des améliorations significativement supérieures par rapport aux valeurs initiales ont été observées dans le psoriasis des ongles (mesuré par le Nail Psoriasis Severity Index [NAPSI]), dans le psoriasis du cuir chevelu (mesuré par le Psoriasis Scalp Severity Index [PSSI]) et le psoriasis palmoplantaire (mesuré par le Psoriasis Palmoplantar Severity Index [PPASI]).
Taltz a été associé à une efficacité précoce avec une diminution > 50 % du score PASI moyen à la semaine 2 (Figure 1). Le pourcentage de patients ayant obtenu une réponse PASI 75 était significativement supérieur dans le groupe Taltz comparé aux groupes placebo et étanercept dès la semaine 1 dans les trois études. Environ 25 % des patients traités par Taltz ont obtenu un score PASI < 5 à la semaine 2, plus de 55 % ont obtenu un score PASI < 5 à la semaine 4 et jusqu'à 85 % à la semaine 12 (contre respectivement 3 %, 14 % et 50 % dans le groupe étanercept). Une amélioration significative de l'intensité des démangeaisons a été observée à la semaine 1 chez les patients traités par Taltz.
Taltz a également montré un effet favorable sur le prurit, qui était significativement supérieur au placebo.
Figure 1. Score PASI, amélioration en pourcentage à chaque visite suivant la visite initiale (mBOCF) dans la population en intention de traiter pendant la phase d'induction (UNCOVER-2 et UNCOVER-3)

L'efficacité et la sécurité de Taltz ont été démontrées quels que soient l'âge, le sexe, l'origine ethnique, le poids corporel, le score de gravité PASI initial, et la prise d'un traitement antérieur biologique. Les taux de réponse après 12 semaines chez les patients traités par Taltz 80 mg 1x/2 sem étaient pour les sous-groupes < 100 kg et ≥100kg respectivement PASI 75 (90.1% et 85.7%) et sPGA 0,1 (84.5% et 75.6%). Les taux de réponse après 12 semaines chez les patients traités par Taltz 80 mg 1x/4 sem étaient pour les sous-groupes < 100 kg et ≥100kg respectivement PASI 75 (85.6% et 74.2%) et sPGA 0,1 (79.0% et 67.4%). La réponse n'a pas été différente chez les patients avec psoriasis des ongles, psoriasis du visage ou psoriasis du cuir chevelu au début de l'étude. Taltz s'est montré efficace chez les patients ayant reçu précédemment un traitement systémique, avec ou sans exposition biologique/anti-TNF préalable, y compris des patients en échec aux traitements biologiques/anti-TNF. Les améliorations des critères d'évaluation sPGA et PASI chez les patients présentant une arthrite psoriasique concomitante au départ de l'étude étaient semblables à ceux de l'ensemble de la population avec psoriasis en plaques modéré à sévère. Environ 45% des patients présentaient un psoriasis dans la zone du visage au départ de l'étude. Parmi ces patients, 80,4% des patients traités avec Taltz étaient débarrassées du psoriasis dans la zone du visage à la semaine 12.
Efficacité chez les non-répondeurs à l'étanercept: chez les patients identifiés comme non-répondeurs à l'étanercept selon le score sPGA (0,1) à la semaine 12 dans l'étude UNCOVER-2 (N = 200) puis traités par Taltz 80 mg 1x/4 sem. après une période de sevrage thérapeutique de 4 semaines, 73 % ont obtenu un score sPGA (0,1) et 83.5 % un PASI 75 après 12 semaines de traitement.
Maintien de la réponse à la semaine 60
Afin d'évaluer le maintien de la réponse, les patients initialement randomisés dans le groupe Taltz et considérés comme répondeurs à la semaine 12 (c'est-à-dire, score sPGA de 0,1) dans les études UNCOVER-1 et UNCOVER-2 ont été re-randomisés pour 48 semaines supplémentaires dans l'un des groupes de traitement suivants: placebo ou Taltz (80 mg toutes les quatre ou douze semaines ([1x/4 sem. ou 1x/12 sem.]).
Les patients qui n'ont pas atteint un sPGA (0,1) à la semaine 12 ou qui ont subi une rechute pendant la phase de maintenance (sPGA ≥3), ont reçu par la suite Taltz 1x/4 sem.
Chez les répondeurs sPGA (0,1) à la semaine 12 (dans les études combinées UNCOVER-1 et UNCOVER-2), la proportion de patients qui ont maintenu cette réponse à la semaine 60 était significativement plus élevée chez les patients traités avec Taltz 80 mg 1x/4 sem. (71 %) comparé à ceux traités par Taltz 80 mg 1x/12 sem. (35.5 %) ou le placebo (7 %).
Le tableau 2 montre les taux de réponse chez les patients re-randomisés pour recevoir la dose de maintenance recommandée de Taltz 80 mg toutes les 4 semaines, en fonction de la dose randomisée à l'inclusion.
Tableau 2. Maintien de la réponse et de l'efficacité au bout de 60 semaines (résultats combinés des études UNCOVER-1 et UNCOVER-2, NRI)
Critère d'évaluation | 80 mg 1x/2 sem. (induction) / Placebo (maintien) (N = 211) | 80 mg 1x/4 sem. (induction) / Placebo (maintien) (N = 191) | 80 mg 1x/2 sem (induction) / 80 mg 1x/4 sem (maintien) (N = 221) | 80 mg 1x/4 sem. (induction) / 80 mg 1x/4 sem. (maintien) (N = 195) |
|---|---|---|---|---|
Score sPGA de «0» (blanchi) ou de «1» (minimal) conservé | 7.6 % | 6.3 % | 78.3 % | 68.7 % |
Score sPGA de «0» (blanchi) conservé ou atteint | 2.8 % | 1.6 % | 58.8 % | 49.2 % |
PASI 75 conservé ou atteint | 9.0 % | 7.9 % | 83.3 % | 74.4 % |
PASI 90 conservé ou atteint t | 4.7 % | 4.7 % | 76.5 % | 66.7 % |
PASI 100 conservé ou atteint t | 2.8 % | 1.6 % | 57.5 % | 49.7 % |
Abréviations: N = nombre de patients dans la population d'analyse; NRI = Non-Responder Imputation
76.4% des répondeurs sPGA (0,1) à la semaine 12, qui ont été assignés au traitement de maintenance Taltz 80 mg 1x/4 sem., avaient un PASI <5 maintenu ou atteint à la semaine 60. L'amélioration de la gravité de la démangeaison a été maintenue jusqu'à la semaine 60 chez les patients traités avec Taltz qui étaient répondeurs sPGA (0,1) à la semaine 12. En ce qui concerne le maintien de la réponse jusqu'à la semaine 60, Taltz a permis de maintenir la réponse chez les patients ayant précédemment reçu un traitement systémique, avec ou sans exposition préalable à des traitements biologiques/anti-TNF, y compris les patients en échec aux traitements biologiques/anti-TNF.
Pour les patients ayant obtenu un score sPGA de répondeurs (0,1) à la semaine 12 et re-randomisés dans le groupe placebo, le délai moyen de rechute (sPGA ≥3) a été de 164 (95% CI [143, 169]) jours en regroupant les études UNCOVER-1 et UNCOVER-2. Parmi ces patients, 71.5 % ont de nouveau obtenu un score sPGA de 0,1 dans les 12 semaines suivant la reprise du traitement par Taltz 80 mg 1x/4 sem.
Chez les patients ayant obtenu un score sPGA de répondeurs (0,1) à la semaine 12, les améliorations du psoriasis des ongles, du cuir chevelu et palmoplantaire persistaient aussi jusqu'à la semaine 60.
Qualité de vie/Résultats rapportés par les patients
Des améliorations statistiquement significatives du score DLQI (Dermatology Life Quality Index ou indice de qualité de vie en dermatologie) ont été démontrées entre l'inclusion et la 12e semaine (études 1 à 3). Ces améliorations se sont maintenues pendant 60 semaines. Taltz était associé à des améliorations significativement supérieures des douleurs cutanées (mesurées au moyen de l'échelle analogique visuelle, visual analogic scale ou VAS).
L'efficacité et la sécurité de l'ixekizumab ont également été étudiées dans l'étude en double-aveugle RHBS, en comparaison à l'ustekinumab, avec une supériorité de l'ixekizumab sur le critère principal de l'étude (réponse PASI 90 à la semaine 12, tableau 3). La supériorité versus ustekinumab a été démontrée rapidement dans les 3 catégories de réponse PASI. La supériorité de l'ixekizumab sur l'ustekinumab a également été démontrée dans les sous-groupes stratifiés par poids.
Tableau 3: Taux de réponse PASI dans l'étude comparative ixekizumab versus ustekinumab
Semaine 12 | Semaine 52 | |||
|---|---|---|---|---|
ixekizumab* | ustekinumab** | ixekizumab* | ustekinumab** | |
Patients (n) | 136 | 166 | 136 | 166 |
PASI 75, n (%) | 120 (88.2 %) | 114 (68.7 %) | 120 (88.2 %) | 126 (75.9 %) |
PASI 90, n (%) | 99 (72.8 %)§ | 70 (42.2 %) | 104 (76.5 %) | 98 (59.0 %) |
PASI 100, n (%) | 49 (36.0 %) | 24 (14.5 %) | 71 (52.2 %) | 59 (35.5 %) |
* 160 mg d'ixekizumab a été administrée en dose initiale suivie de 80 mg aux semaines 2, 4, 6, 8, 10 et 12, et 80 mg 1x/4 sem. par la suite
** Posologie en fonction du poids: les patients traités par ustekinumab ont eu une dose de 45 mg ou 90 mg aux semaines 0 et 4, puis toutes les 12 semaines jusqu'à la semaine 52 (dose en fonction du poids conformément à la posologie approuvée)
§ p < 0.001 versus ustekinumab (valeur de p mentionnée uniquement pour le critère principal)
Efficacité dans le psoriasis génital
Une étude randomisée en double-aveugle, contrôlée versus placebo (IXORA-Q) a été conduite chez 149 patients adultes (24 % de femmes) présentant un psoriasis génital modéré à sévère (score du sPGA-Génital ≥3), avec une atteinte d'au moins 1 % de la surface corporelle (body surface area, BSA) (60.4 % avaient une BSA ≥10 %) et un antécédent d'échec ou d'intolérance à au moins un traitement topique pour un psoriasis génital. Les patients présentaient un psoriasis en plaques au-moins modéré (défini par un score sPGA ≥3 et candidats à une photothérapie et/ou à un traitement systémique) depuis au moins 6 mois.
Les participants à l'étude randomisés dans le groupe Taltz ont reçu une dose initiale de 160 mg suivie de 80 mg toutes les 2 semaines pendant 12 semaines. Le critère principal était la proportion de patients ayant atteint une réponse du sPGA-Génital de «0» (blanchi) ou «1» (minimal) (sPGA-G 0/1). A la semaine 12, il y a eu significativement plus de patients dans le groupe Taltz comparé au groupe placebo qui ont atteint un sPGA-G 0/1 et un sPGA 0/1 indépendamment de la surface corporelle à l'inclusion (surface corporelle à l'inclusion BSA 1 % à <10 %, resp. BSA ≥10 %: réponse sPGA-G de «0» ou «1»: Taltz 71 % resp. 75 %; placebo 0 % resp. 13 %).
Psoriasis en plaques pédiatrique
Une étude clinique multicentrique, randomisée, en double-aveugle, contrôlée contre placebo (IXORA‑Peds) a inclus 201 patients pédiatriques âgés de 6 ans à moins de 18 ans, atteints de psoriasis en plaques modéré à sévère (défini par un score sPGA ≥3 avec une atteinte ≥10 % de la surface corporelle et un score PASI ≥12) qui étaient candidats à la photothérapie ou à un traitement systémique, ou qui étaient insuffisamment contrôlés par un traitement topique.
Les principaux critères d'exclusion étaient similaires à ceux des études mentionnées plus haut chez l'adulte, en particulier les infections sévères actives ou récentes ainsi que les traitements concomitants du psoriasis systémiques conventionnels ou biologiques ou la photothérapie.
Durant la phase de 12 semaines en double aveugle contrôlée contre placebo et la phase de contrôle actif, les patients ont été traités par placebo (n = 56), étanercept (n = 30) ou Taltz (n = 115), la posologie adaptée au poids étant ajustée comme suit:
Taltz: <25 kg: 40 mg à la semaine 0, suivi de 20 mg 1x/4 sem.
de 25 kg à 50 kg: 80 mg à la semaine 0, suivi de 40 mg 1x/4 sem.
>50 kg: 160 mg à la semaine 0, suivi de 80 mg 1x/4 sem.
Etanecerpt: 0.8 mg/kg, maximum 50 mg par dose 1x/ sem.
La réponse au traitement a été évaluée après 12 semaines de traitement et a été définie par la proportion de patients ayant atteint le co‑critère d'évaluation principal, par un score sPGA de «0» (blanchi) ou «1» (minimal) avec une amélioration d'au moins 2 points par rapport au score à l'inclusion, ainsi que par la proportion de patients ayant atteint une réduction du score PASI d'au moins 75 % (PASI 75) par rapport au score à l'inclusion.
Les autres résultats évalués à la semaine 12 comprenaient la proportion de patients ayant atteint un score PASI 90, PASI 100, sPGA de «0» et une amélioration de la sévérité des démangeaisons, mesurée par une réduction d'au moins 4 points sur une échelle d'évaluation numérique des démangeaisons de 11 points (itch Numeric Rating Scale, itch NRS).
Les patients avaient un score PASI médian de 17 à l'inclusion, les scores allant de 12 à 49. Le score sPGA à l'inclusion était sévère ou très sévère dans 49 % des cas. Sur l'ensemble des patients, 22 % avaient préalablement reçu une photothérapie, 32 % un traitement systémique conventionnel et 4% un traitement biologique pour le traitement du psoriasis.
Les données de réponse clinique sont présentées dans le Tableau 4.
Tableau 4: Résultats d'efficacité à 12 semaines chez les patients pédiatriques avec psoriasis en plaques, NRI
Critères d'évaluation | Taltza (N=115) n (%) | Placebo (N=56) n (%) | Différence vs placebo (95% CI) |
|---|---|---|---|
sPGA «0» (blanchi) ou «1» (minimal)b | 93 (81) | 6 (11) | 70.2 (59.3, 81.0) d |
sPGA «0» (blanchi) | 60 (52) | 1 (2) | 50.4 (40.6, 60.2) d |
PASI 75c | 102 (89) | 14 (25) | 63.7 (51.0, 76.4)d |
PASI 90 | 90 (78) | 3 (5) | 72.9 (63.3, 82.5)d |
PASI 100 | 57 (50) | 1 (2) | 47.8 (38.0, 57.6)d |
Score Itch NRS (amélioration ≥4 points) c | 59 (71) | 8 (20) | 51.1 (35.3, 66.9) d |
Abréviations: N = Nombre de patients dans la population en intention de traiter (ITT); NRI = Non-Responder Imputation ou imputation d'absence de réponse.
a À la semaine 0, les patients ont reçu une dose de 160 mg (poids coroporel [PC] >50kg),, 80 mg (PC 25–50 kg), ou de 40 mg (PC < 50 kg) de Taltz, suivie de 80 mg (PC >50kg), 40 mg (PC 25–50 kg), ou 20 mg (< 25 kg) toutes les 4 semaines pendant 12 semaines.
b Co-critères d'évaluation principaux.
c Score Itch NRS (amélioration ≥4) chez les patients ayant un score Itch NRS ≥4 à l'inclusion. Le nombre de patients en ITT avec un score Itch NRS ≥4 à l'inclusion est le suivant: Taltz, n = 83; PBO, n = 40.
d p<0.001
Un total de 87 patients pédiatriques atteints de psoriasis en plaques sévère (PASI ≥20 ou sPGA ≥4) ont été randomisés pour recevoir de l'ixekizumab 1x/4 sem. (38 patients), de l'étanercept 1x/ sem. (30 patients) ou un placebo (19 patients).
À la semaine 12, des améliorations ont été observées pour le groupe ixekizumab 1x/4 sem. par rapport au groupe étanercept 1x/ sem. et au groupe placebo, mesurées par PASI 75 (84.2%, 63.3%, 26.3%) et sPGA (0,1) (76.3%, 53.3% und 5.3%).
Les critères secondaires les plus importants ont montré des améliorations statistiquement significatives dans le groupe ixekizumab Q4W par rapport au groupe étanercept Q1W et au groupe placebo, mesurées par: PASI 90 (76.3%, 40.0%, 0), PASI 100 (60.5%, 16.7%, 0) et sPGA (0) (63.2%, 16.7%, 0).
Les patients du groupe de traitement ixékizumab ont présenté une réponse CDLQI (children Dermatology Life Quality Index)/DLQI (0,1) à la semaine 12 (NRI) plus élevée comparé au placebo. La différence entre les groupes de traitement était observée dès la semaine 4.
Il n'y a pas de données après l'arrêt de Taltz chez les enfants et sur le risque de poussée de la maladie.
Arthrite psoriasique
La sécurité et l'efficacité de Taltz ont été évaluées dans deux études de phase III randomisées, en double-aveugle, contrôlées versus placebo chez 780 patients atteints d'arthrite psoriasique active (≥3 articulations gonflées et ≥3 articulations douloureuses). Le diagnostic d'arthrite psoriasique (selon les critères de classification de l'arthrite psoriasique CASPAR, Classification Criteria for Psoriatic Arthritis) a été posé chez les patients dans une médiane de 5.33 ans avant l'inclusion dans l'étude. Les patients randomisés présentaient des lésions cutanées de psoriasis en plaques actuelles (94.0 %) ou des antécédents documentés de psoriasis en plaques. A l'inclusion, 12.1 % des patients étaient atteints de psoriasis en plaques modéré à sévère. A l'inclusion, plus de 58.9 % des patients atteints d'arthrite psoriasique avaient des enthésites et 22.3 % des dactylites. Dans les deux études, le critère principal était le taux de réponse American College of Rheumatology (ACR) 20 à la semaine 24.
Dans l'étude 1 dans l'arthrite psoriasique (SPIRIT-P1), des patients atteints d'arthrite psoriasique active naïfs de traitement biologique ont été randomisés dans les groupes de traitement suivants: injections sous-cutanées de placebo, adalimumab 40 mg toutes les 2 semaines (groupe de contrôle actif), Taltz 80 mg toutes les 2 semaines (1x/2 sem.) ou Taltz 80 mg toutes les 4 semaines (1x/4 sem.). Les deux schémas posologiques de Taltz comprenaient une dose initiale était de 160 mg. Dans cette étude, 85.3 % des patients avaient déjà été traités par au moins un DMARD conventionnel. 53 % des patients étaient traités de façon concomitante par MTX à une dose hebdomadaire moyenne de 15.8 mg. 67 % des patients qui étaient traités de façon concomitante par MTX recevaient une dose supérieure ou égale à 15 mg. Dans tous les groupes de traitement, les patients ayant une réponse insuffisante à la semaine 16 ont reçu un traitement de secours (modification du traitement de fond). Les patients traités par Taltz 1x/2 sem. ou 1x/4 sem. ont conservé la dose initiale de Taltz. Les patients recevant adalimumab ou le placebo ont été re-randomisés selon un rapport 1:1 pour recevoir Taltz 1x/2 sem. ou 1x/4 sem. à la semaine 16 ou 24 en fonction du statut de répondeur.
L'étude 2 dans l'arthrite psoriasique (SPIRIT-P2) incluait des patients ayant déjà été traités par un anti-TNF et ayant arrêté le traitement par anti-TNF en raison d'un manque d'efficacité ou d'une intolérance (patients anti-TNF-IR). Les patients ont été randomisés dans les groupes de traitement suivants: injections sous-cutanées de placebo, Taltz 80 mg toutes les 2 semaines (1x/2 sem.) ou Taltz 80 mg toutes les 4 semaines (1x/4 sem.). Les deux schémas posologiques de Taltz comprenaient une dose initiale était de 160 mg. 56 % et 35 % des patients ne répondaient pas de façon adéquate à respectivement 1 anti-TNF et ≥2 anti-TNF. L'étude SPIRIT-P2 a évalué 363 patients, parmi lesquels 41 % recevaient un traitement concomitant par MTX à une dose hebdomadaire moyenne de 16.1 mg. 73.2 % des patients traités de façon concomitante par MTX avaient une dose supérieure ou égale à 15 mg. Dans tous les groupes de traitement, les patients ayant eu une réponse insuffisante à la semaine 16 ont reçu un traitement de secours (modification du traitement de fond). Les patients traités par Taltz 1x/2 sem. ou 1x/4 sem. ont conservé la dose initiale de Taltz. Les patients recevant le placebo ont été re-randomisés selon un rapport 1:1 pour recevoir Taltz 1x/2 sem. ou 1x/4 sem. à la semaine 16 ou 24 en fonction du statut de répondeur.
Signes et symptômes
Le traitement par Taltz a montré une amélioration significative des mesures de l'activité de la maladie par rapport au placebo à la semaine 24 (voir Tableau 5.
Tableau 5 Résultats d'efficacité des études SPIRIT-P1 et SPIRIT-P2 à la semaine 24
SPIRIT-P1 | SPIRIT-P2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Critères d'évalua-tion | Différence de taux de réponse versus placebo (CI 95 %) | Différence de taux de réponse versus placebo (CI 95 %) | ||||||||
PBO (N = 106) | Taltz 1x/4 sem. (N = 107) | Taltz 1x/2sem. (N = 103) | Taltz 1x/4 sem. | Taltz 1x/2 sem. | PBO (N = 118) | Taltz 1x/4 sem. (N = 122) | Taltz 1x/2 sem. (N = 123) | Taltz 1x/4 sem. | Taltz 1x/2 sem. | |
Taux de réponse ACR 20, n (%) | ||||||||||
Semaine 24 | 32 (30.2) | 62 (57.9) | 64 (62.1) | 27,8 (15.0; 40.6)* | 31,9 (19.1; 44.8)* | 23 (19.5) | 65 (53.3) | 59 (48.0) | 33.8 (22.4; 45.2)* | 28.5 (17.1; 39.8)* |
Taux de réponse ACR 50, n (%) | ||||||||||
Semaine 24 | 16 (15.1) | 43 (40.2) | 48 (46.6) | 25.1 (13.6; 36.6)* | 31.5 (19.7; 43.3)* | 6 (5.1) | 43 (35.2) | 41 (33.3) | 30.2 (20.8; 39.5)* | 28.3 (19.0; 37.5)* |
Taux de réponse ACR 70, n (%) | ||||||||||
Semaine 24 | 6 (5.7) | 25 (23.4) | 35 (34.0) | 17.7 (8.6; 26.8)* | 28.3 (18.2; 38.5)* | 0 | 27 (22.1) | 15 (12.2) | 22.1 (14.8; 29.5)* | 12.2 (6.4; 18.0)* |
Abréviations: ACR 20/50/70 = Taux de réponse de 20 %/50 %/70 % de l'American College of Rheumatology; CI = intervalle de confiance; 1x/4 sem. = Taltz 80 mg une fois toutes les 4 semaines; 1x/2 sem. = Taltz 80 mg une fois toutes les 2 semaines; N = nombre de patients dans la population analysée; n = nombre de patients dans une catégorie précise; NRI = non-responder imputation; PBO = placebo.
Note: les patients ayant reçu un traitement de secours à la semaine 16 ou ayant arrêté le traitement ou pour lesquels des données manquaient ont été imputés comme non-répondeurs pour les analyses à la semaine 24.
Les DMARDs conventionnels concomitants incluaient MTX, léflunomide et sulfasalazine.
* p<0.001 par rapport au placebo.
Chez les patients avec des dactylites ou des enthésites pré-existantes, le traitement par Taltz 1x/4 sem. a entraîné une amélioration des dactylites et des enthésites à la semaine 24 comparé au placebo (p<0.01).
Chez les patients atteints à la fois d'un psoriasis en plaques (≥3% de surface corporelle atteinte) et d'arthrite psoriasique, le traitement avec Taltz 1x/4 sem. a entraîné une amélioration des lésions cutanées psoriatiques, comme démontré par les réponses PASI 75, PASI 90 et PASI 100 à la semaine 24 (p<0.001).
Chez les patients atteints à la fois d'un psoriasis en plaques modéré à sévère et d'arthrite psoriasique, le schéma posologique de Taltz 1x/2 sem. a montré un taux de réponse significativement plus élevé pour PASI 75, PASI 90 et PASI 100 par rapport au placebo (p<0.001) et a démontré un bénéfice cliniquement significatif par rapport au schéma posologique de Taltz 1x/4 sem.
Les réponses au traitement avec Taltz étaient significativement plus élevées que celles avec placebo dès la semaine 1 pour l'ACR 20, dès la semaine 4 pour l'ACR 50 et dès la semaine 8 pour l'ACR 70, et ces réponses ont persisté jusqu'à la semaine 24.
Figure 2: Taux de réponse ACR 20 dans l'étude SPIRIT-P1 jusqu'à la semaine 24

Pour Taltz 1x/2 sem. et 1x/4 sem.: b p<0.01 et c p<0.001 par rapport au placebo.
Dans SPIRIT-P1 et SPIRIT-P2, des taux de réponses similaires pour les ACR 20/50/70 ont été observés chez les patients atteints d'arthrite psoriasique indépendamment du fait qu'ils recevaient ou non un traitement concomitant par cDMARDs, y compris MTX.
Dans SPIRIT-P1 et SPIRIT-P2, des améliorations ont été observées pour toutes les composantes des scores ACR, y compris pour le critère d'évaluation de la douleur par le patient.
Dans SPIRIT-P1, l'efficacité a été maintenue jusqu'à la semaine 52 comme démontré par les taux de réponses de l'ACR 20/50/70, l'amélioration des enthésites, l'amélioration des dactilytes et le PASI 75/90/100.
L'efficacité et la sécurité de Taltz ont été démontrées quels que soient l'âge, le sexe, l'origine ethnique, la durée de la maladie, le poids corporel à l'inclusion dans l'étude, la présence de psoriasis à l'inclusion dans l'étude, la CRP initiale, la DAS28-CRP initiale, la prise concomitante de corticoïdes et un traitement antérieur par un biologique. Taltz a été efficace chez les patients naïfs de traitement biologique, ayant déjà été exposés à un traitement biologique et n'ayant pas répondu à un traitement biologique.
Il y avait trop peu de patients atteints d'arthrite mutilante, d'arthrite isolée avec atteinte des articulations interphalangiennes distales (IPD) et de spondylarthrite concomitante dans les études d'homologation pour obtenir des résultats significatifs dans ces sous-groupes spécifiques.
Réponse radiographique
Dans SPIRIT-P1, l'inhibition de la progression des atteintes structurales articulaires a été évaluée par radiographie et exprimée au moyen du Score total de Sharp modifié (mTSS) et de ses composants, le score d'érosion (ES, Erosion Score) et le score de pincement articulaire (JSN, Joint Space Narrowing) aux semaines 24 et 52, par rapport aux valeurs à l'inclusion.
La progression des atteintes articulaires radiographiques (mTSS) était inhibée par Taltz à la semaine 24 par rapport au placebo.
Le pourcentage de patients ne présentant pas de progression des atteintes articulaires radiographiques (définie par une variation du mTSS par rapport à l'inclusion ≤0.5) entre la randomisation et la semaine 24 était de 94.8 % chez les patients traités par Taltz 1x/2 sem., 89.0 % avec Taltz 1x/4 sem. et 77.4 % avec le placebo. L'inhibition des atteintes structurales a été maintenue avec le traitement par Taltz jusqu'à la semaine 52.
Capacité fonctionnelle et qualité de vie liée à la santé
Dans SPIRIT-P1 et SPIRIT-P2, les patients traités par Taltz 1x/2 sem. (p<0.001) et 1x/4 sem. (p<0.001) ont présenté une amélioration significative de leur capacité fonctionnelle, évaluée à l'aide de l'indice HAQ-DI (Health Assessment Questionnaire – Disability Index) à la semaine 24 et maintenue à la semaine 52 dans l'étude SPIRIT-P1.
Les patients traités par Taltz ont rapporté des améliorations de la qualité de vie liée à la santé, évaluée par le score Physical Component Summary of the Short Form-36 Health Survey (SF‑36 PCS) (p<0.001). Une amélioration statistiquement significative de la fatigue a aussi été démontrée au moyen de l'échelle Fatigue Severity NRS.
Spondylarthrite ankylosante (spondyloarthrite axiale radiographique, maladie de Bechterew)
La sécurité et l'efficacité de Taltz ont été étudiées dans 2 études randomisées, en double-aveugle, contrôlées contre placebo (COAST-V et COAST-W) chez 657 patients adultes ≥18 ans atteints de spondylarthrite ankylosante. Les patients avaient une maladie active définie selon le Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 et des douleurs rachidiennes ≥4 sur une échelle d'évaluation numérique malgré le traitement par AINS. Dans les deux études, les patients présentaient à l'inclusion des symptômes de spondylarthrite ankylosante depuis en moyenne 17 ans (médiane de 16 ans). A l'inclusion, environ 32 % des patients recevaient un traitement concomitant par cDMARD (dans les études COAST-V et COAST-W: méthotrexate 8.5 % et 13.0 %, sulfasalazine 28.5 % et 14.6 %, hydroxychloroquine 0.6 % et 0%).
Dans les deux études, le critère d'évaluation principal était le pourcentage de patients ayant obtenu une réponse Assessment of Spondyloarthritis International Society 40 (ASAS40) à la semaine 16.
COAST-V a évalué 341 patients naïfs de traitement biologique traités avec Taltz 80 mg ou 160 mg à la semaine 0, suivis de 80 mg toutes les 2 semaines (1x/2 sem.) ou 4 semaines (1x/4 sem), adalimumab 40 mg toutes les 2 semaines ou placebo. Les patients traités avec le placebo ont été re-randomisés à la semaine 16 pour recevoir Taltz (dose initiale de 160 mg suivie de 80 mg 1x/2 sem. ou 1x/4 sem). Les patients traités avec adalimumab ont été re-randomisés à la semaine 16 pour recevoir Taltz (80 mg 1x/2 sem. ou 1x/4 sem.).
COAST-W a évalué 316 patients précédemment traités avec 1 ou 2 inhibiteurs du TNF (90 % ayant eu une réponse inadéquate et 10 % une intolérance aux inhibiteurs du TNF). Tous les patients ont été traités avec Taltz 80 mg ou 160 mg à la semaine 0, puis avec 80 mg 1x/2 sem. ou 1x/4 sem. ou avec placebo. Les patients traités avec le placebo ont été re-randomisés à la semaine 16 pour recevoir Taltz (dose initiale de 160 mg suivie de 80 mg 1x/2 sem. ou 1x/4 sem.).
Réponse clinique
Dans les deux études, les patients traités par Taltz 80 mg 1x/2 sem. ou 1x/4 sem. ont présenté des améliorations plus importantes de la réponse ASAS40 et ASAS20 par rapport au placebo à la semaine 16 (tableau 6). La réponse était comparable chez les patients, indépendamment des thérapies concomitantes. Dans COAST-W, la réponse était indépendante du nombre de traitements antérieurs par des inhibiteurs du TNF.
Tableau 6 Résultats d'efficacité des études COAST-V et COAST-W à la semaine 16
COAST-V, naïfs de traitement biologique | COAST-W, expérience avec inhibiteurs du TNF | ||||||
|---|---|---|---|---|---|---|---|
Taltz 80 mg 1x/4 sem.a (N=81) | Placebo (N=87) | Différence par rapport au placebo (CI 95%) | Adalimumab 40 mg 1x/2 sem. (N= 90) | Taltz 80 mg 1x/4 sem.c (N=114) | Placebo (N=104) | Différence par rapport au placebo (CI 95%) | |
Réponse ASAS20b, n (%), NRI | 52 (64.2%) | 35 (40.2%) | 24.0 (9.3, 38.6)** | 53 (58.9%) | 55 (48.2%) | 31 (29.8%) | 18.4 (5.7, 31.1)** |
Réponse ASAS40b,c, n (%), NRI | 39 (48.1%) | 16 (18.4%) | 29.8 (16.2, 43.3)*** | 32 (35.6%) | 29 (25.4%) | 13 (12.5%) | 12.9 (2.7, 23.2)* |
ASDAS à l'inclusion[variation vs inclusion] | 3.7 | 3.9 | -1.0 (-1.3, -0.7)*** | 3.7 | 4.2 [‑1.2] | 4.1 | -1.1 (-1.3, -0.8)*** |
Score BASDAI à l'inclusion [variation vs inclusion] | 6.8 [-2.9] | 6.8 [-1.4] | -1.5 (-2.1, -0.9)*** | 6.7 [-2.5]*** | 7.5 [‑2.2] | 7.3 [-0.9] | -1.2 (-1.8, -0.7)*** |
MRI Spine SPARCCd à l'inclusion [variation vs inclusion] | 14.5 [‑11.0] | 15.8 | -9.5 (-12.6, -6.4)*** | 20.0 | 8.3 [‑3.0] | 6.4 | -6.3 (-10.0, -2.5)** |
BASDAI50e (%), NRI | 42 | 17 | 25 (11,38)*** | 32* | 22 | 10 | 12 (3, 22)* |
ASDAS <2.1 (%) (faible activité de la maladie), NRI | 43 | 13 | 31 (18,43)*** | 38*** | 18 | 5 | 13 (5, 21)** |
ASAS HI à l'inclusion [variation vs inclusion] | 7.5 | 8.1 | -1.1 (-2.0, -0.3)* | 8.2 | 10.0 [‑1.9] | 9.0 | -1.0 (-1.9, -0.1)* |
SF-36 PCS à l'inclusion [variation vs inclusion] | 34.0 [7.7] | 32.0 [3.6] | 4.1 (1.9, 6.2)*** | 33.5 [6.9]** | 27.5 [6.6]*** | 30.6 [1.4] | 5.2 (3.0, 7.4)*** |
Abréviations: N = nombre de patients dans la population en intention de traiter; NRI = Non-Responder Imputation ou imputation d'absence de réponse; les patients ayant des données manquantes ont été comptés comme non-répondeurs.
ASAS HI = Assessment of SpondyloArthritis International Society Health Index, Indice d'évaluation de la santé dans la spondyloarthrite; ASDAS = Ankylosing Spondylitis Disease Activity Score, Score d'activité de la spondylarthrite ankylosante; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index, Indice d'activité de la spondylarthrite ankylosante de Bath; Variation par rapport à l'inclusion = modification de la moyenne des moindres carrés à la semaine 16 par rapport à la valeur à l'inclusion; MRI Spine SPARCC = Spondyloarthritis Research Consortium of Canada Magnetic Resonance Imaging Scoring of the Spine, Score IRM-rachis du Consortium canadien de recherche sur la spondyloarthrite (cotation de 23 unités discovertébrales) SF-36 PCS = Short Form health survey physical component summary.
a À la semaine 0, les patients ont reçu Taltz 80 mg ou 160 mg..
b Une réponse ASAS20 est définie comme une amélioration ≥20 % et une amélioration absolue par rapport à la valeur à l'inclusion ≥1 unité (intervalle de 0 à 10) dans au moins 3 des 4 domaines (Évaluation globale du patient, douleur rachidienne, fonction et inflammation), et aucune aggravation ≥20 % et ≥1 unité (intervalle de 0 à 10) dans le domaine restant. Une réponse ASAS40 est définie comme une amélioration ≥40 % et une amélioration absolue par rapport à la valeur à l'inclusion ≥2 unités dans au moins 3 des 4 domaines, sans aucune aggravation dans le domaine restant.
c Critère principal.
d Les nombres de patients en ITT avec des données IRM à l'inclusion sont les suivants: COAST-V: Taltz, n = 81; PBO, n = 82; ADA, n = 85. COAST-W: Taltz, n = 58; PBO, n = 51.
e Réponse BASDAI50 définie comme une amélioration ≥50 % du score BASDAI par rapport à sa valeur à l'inclusion.
* p<0.05; ** p<0.01; *** p<0.001 par rapport au placebo.
L'amélioration des principaux composants du critère de réponse ASAS40 (douleurs rachidiennes, BASFI, évaluation globale du patient, raideur) et d'autres mesures d'activité de la maladie, y compris la CRP, a démontré un bénéfice clinique significatif à la semaine 16.
Le pourcentage de patients ayant atteint une réponse ASAS40 dans les études COAST-V et COAST-W est présenté dans la figure 3.
Figure 3. Réponse ASAS40 dans les études COAST-V et COAST-W jusqu'à la semaine 16, NRIa

a Les patients ayant des données manquantes ont été comptés comme non-répondeurs.
* p<0.05; ** p<0.01; *** p<0.001 par rapport au placebo
Un taux de réponse ASAS40 comparable a été observé chez les patients indépendamment des taux de CRP à l'inclusion, des scores ASDAS à l'inclusion et des scores IRM SPARCC-rachis. La réponse ASAS40 a été démontrée indépendamment de l'âge, du sexe, de l'ethnie, de la durée de la maladie, du poids corporel à l'inclusion, du score BASDAI à l'inclusion et du traitement biologique antérieur.
Dans les études COAST-V et COAST-W, l'efficacité a été maintenue jusqu'à la semaine 52, selon les critères d'évaluation présentés ci-dessus à la semaine 16, y compris les taux de réponse ASAS20, ASAS40, ASDAS, BASDAI, BASFI et ASAS HI.
Résultats liés à la santé
Des améliorations des douleurs rachidiennes par rapport au placebo ont été observées dès la semaine 1, et se sont maintenues jusqu'à la semaine 16 (Taltz vs placebo: COAST-V ‑3.2 vs -1.7; COAST-W -2.4 vs -1.0); des améliorations sur la fatigue et sur la mobilité rachidienne ont été observées par rapport au placebo à la semaine 16. Les améliorations sur la douleur rachidienne, sur la fatigue et sur la mobilité rachidienne se sont maintenues jusqu'à la semaine 52.
Pharmacocinétique
Les propriétés pharmacocinétiques de Taltz étaient comparables dans les indications psoriasis en plaques, arthrite psoriasique et spondylarthrite ankylosante.
Absorption
Après une dose unique sous-cutanée d'ixekizumab chez des patients atteints de psoriasis, les concentrations sériques maximales moyennes ont été obtenues dans les 4 à 7 jours, avec un intervalle de dose compris entre 5 et 160 mg. La concentration sérique maximale (Cmax) moyenne (écart-type) d'ixekizumab, après une dose initiale de 160 mg, a été de 19.9 (8.15) µg/ml.
Après une dose initiale de 160 mg, l'état d'équilibre a été obtenu à la semaine 8 avec la posologie de 80 mg 1x/2 sem. Les estimations de la Cmax,ss, (concentration maximale à l'état d'équilibre) et de la Ctrough,ss (concentration résiduelle à l'état d'équilibre) moyennes (écart-type) sont respectivement de 21.5 (9.16) µg/mL et de 5.23 (3.19) µg/mL.
Après le passage à la semaine 12, du régime posologique de 80 mg 1x/2 sem. au régime posologique de 80 mg 1x/4 sem., l'état d'équilibre serait atteint au bout de 10 semaines environ. Les estimations de la Cmax,ss, et de la C trough,ss moyennes (écart-type) sont respectivement de 14.6 (6.04) µg/ml et de 1.87 (1.30) µg/ml.
La biodisponibilité absolue moyenne de l'ixekizumab était estimée entre 54 et 90 % selon les analyses. Après injection sous-cutanée dans l'abdomen, le haut du bras ou la cuisse, des concentrations sériques similaires sont atteintes.
Distribution
Selon les analyses pharmacocinétiques de la population, le volume de distribution total moyen à l'état d'équilibre était de 7.11 litres.
Métabolisme
L'ixekizumab étant un anticorps monoclonal, il devrait être décomposé en petits peptides et en acides aminés par les voies cataboliques de la même manière que les IgGs endogènes.
Élimination
Dans l'analyse pharmacocinétique de population, la clairance sérique moyenne a été de 0.0161 l/h. La clairance est indépendante de la dose. La demi-vie d'élimination moyenne, estimée d'après l'analyse pharmacocinétique de la population, est de 13 jours chez les patients atteints de psoriasis en plaques.
Linéarité/non-linéarité
L'exposition (AUC) a augmenté proportionnellement à la dose administrée pour une gamme posologique comprise entre 5 et 160 mg en injection sous-cutanée.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Aucune étude de pharmacologie clinique spécifique destinée à évaluer les effets de l'insuffisance hépatique sur les paramètres pharmacocinétiques de l'ixekizumab n'a été réalisée.
Troubles de la fonction rénale
Aucune étude de pharmacologie clinique spécifique destinée à évaluer les effets de l'insuffisance rénale sur les paramètres pharmacocinétiques de l'ixekizumab n'a été réalisée.
Patients âgés
Sur la base d'une analyse pharmacocinétique de la population avec un nombre limité de patients âgés (n = 94 patients de 65 ans et plus et n = 12 patients de 75 ans et plus), la clairance chez les patients âgés et les patients de moins de 65 ans était similaire.
Enfants et adolescents
Les patients pédiatriques atteints de psoriasis (âgés de 6 ans à moins de 18 ans) ont été traités par ixekizumab pendant 12 semaines au schéma posologique pédiatrique recommandé. Les patients pesant > 50 kg et ceux pesant de 25 à 50 kg avaient une concentration résiduelle moyenne ± écart-type à l'état d'équilibre de 3.8 ± 2.2 µg / ml et 3.9 ± 2.4 µg / ml, respectivement, à la semaine 12. Ces concentrations étaient comparables aux concentrations chez les patients adultes recevant 80 mg d'ixékizumab toutes les 4 semaines, la concentration minimale moyenne ± écart-type à l'état d'équilibre étant de 3.5 ± 2.2 μg / ml.
Données précliniques
Les données non cliniques obtenues chez des singes Cynomolgus n'ont pas révélé de risque particulier pour l'homme sur la base des études de toxicologie en administration répétée, des évaluations de pharmacologie de sécurité et des études de toxicité sur la reproduction et sur le développement.
Toxicité en cas d'administration répétée
L'administration d'ixekizumab à des singes Cynomolgus pendant 39 semaines à des doses sous-cutanées allant jusqu'à 50 mg/kg une fois par semaine n'a produit aucune toxicité d'organe, ni d'effet indésirable sur la fonction immunitaire (par ex., réponse des anticorps dépendant des lymphocytes T et activité des cellules NK). Une dose sous-cutanée hebdomadaire de 50 mg/kg à des singes représente environ 19 fois la dose initiale de 160 mg de Taltz et entraîne, chez les singes, une exposition (AUC) au moins 61 fois supérieure à l'exposition moyenne prévue chez l'homme à l'état d'équilibre après administration de la dose recommandée.
Mutagénicité / Carcinogénicité
Aucune étude non clinique n'a été réalisée pour évaluer le potentiel carcinogène ou mutagène de l'ixekizumab.
Toxicité sur la reproduction
Aucun effet sur les organes reproducteurs, les cycles menstruels ou le sperme n'a été observé chez des singes Cynomolgus sexuellement matures ayant reçu une dose hebdomadaire sous-cutanée de 50 mg/kg d'ixekizumab pendant 13 semaines.
Dans les études de toxicité du développement, l'administration sous-cutanée d'ixekizumab à des doses allant jusqu'à 50 mg/kg une fois par semaine chez des singes Cynomolgus depuis le début de l'organogenèse jusqu'à peu de temps avant la fin de la grossesse ou jusqu'à la naissance n'a pas induit d'effet embryotoxique ou tératogène, et n'a pas eu d'effet sur le cours de l'accouchement ni sur le développement morphologique, fonctionnel ou immunologique de la progéniture de leur naissance jusqu'à l'âge de 6 mois. L'incidence plus élevée de décès postnatal chez les singes était principalement attribuable à la négligence des mères ou à des naissances prématurées. Il s'agit de résultats qui sont fréquemment observés chez les singes et ne sont probablement pas causés par l'ixekizumab. En raison du petit nombre d'animaux, la valeur prédictive de l'étude est limitée. Les études de toxicité du développement ont montré que l'ixekizumab traversait le placenta et était présent dans le sang de la descendance jusqu'à l'âge de 6 mois.
Remarques particulières
Incompatibilités
Non applicable (médicament à dose unique pour utilisation sous-cutanée).
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2–8 °C). Conserver dans l'emballage original, afin de protéger le contenu de la lumière.
Ne pas congeler. Taltz ne doit pas être utilisé s'il a été congelé.
Stockage temporaire: Taltz peut être conservé jusqu'à 5 jours non réfrigéré, à une température inférieure à 30°C.
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Ne pas agiter.
Taltz est une solution stérile, sans agent conservateur, limpide et incolore à légèrement jaune.
Ne pas utiliser si la solution contient des particules ou si elle est trouble et/ou de couleur marron.
Les seringues préremplies et les stylos préremplis sont à usage unique.
Le manuel d'utilisation joint à l'information destinée aux patients de la seringue préremplie, respectivement du stylo prérempli, doit être suivi scrupuleusement.
Préparation de Taltz 40mg pour les patients pédiatriques de 25 kg à 50 kg
Les doses de 40 mg d'ixékizumab doivent être préparées et administrées par un professionnel de santé qualifié. Utilisez uniquement la seringue préremplie Taltz 80 mg/1 ml disponible dans le commerce lorsque vous préparez les doses pédiatriques prescrites de 40 mg.
- injecter tout le contenu de la seringue préremplie dans un flacon stérile en verre transparent. NE PAS secouer ni agiter le flacon.
- utiliser une seringue jetable de 0,5 ml ou 1 ml et une aiguille stérile pour prélever 0.5 ml à partir du flacon.
- changer l'aiguille et utiliser une aiguille stérile de 27 Gauge pour faire l'injection au patient. Jeter l'ixexizumab inutilisé dans le flacon.
La dose préparée d'ixékizumab doit être administrée dans les 4 heures suivant la perforation du flacon stérile à température ambiante.
Numéro d’autorisation
65906, 65907 (Swissmedic).
Titulaire de l’autorisation
Eli Lilly (Suisse) SA, 1214 Vernier/GE.
Mise à jour de l’information
Juillet 2020
Отзывов (0)
Вы смотрели


Бесплатная консультация опытного специалиста
Опишите симптомы или нужный препарат – мы поможем подобрать его дозировку или аналог, оформим заказ с доставкой на дом или просто проконсультируем.
Нас 14 специалистов и 0 ботов. Мы всегда будем с вами на связи и сможем связаться в любое время.