
Nuwiq сухое вещество 250 Ie C растворитель флакон 2,5 мл
NUWIQ Trockensub 250 IE c Solv
-
57629.56 RUB

- Наличие: Нет в наличии
- Модель: 6708094
- ATC-код B02BD02
- EAN 7680655510012
Состав:
Наведите телефон на qr-код

Описание
 Deutsch
Deutsch French
French Italian
ItalianZusammensetzung
Wirkstoffe
Simoctocog alfa (rekombinanter humaner Blutgerinnungsfaktor VIII).
Hilfsstoffe
Saccharose, Natriumchlorid, Calciumchlorid-Dihydrat, Argininhydrochlorid, Natriumcitrat-Dihydrat, Poloxamer 188 (entspricht einem Gesamtnatriumgehalt von 18,4 mg/vial).
Lösungsmittel: Wasser für Injektionszwecke.
Darreichungsform und Wirkstoffmenge pro Einheit
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung.
Jede Durchstechflasche enthält nominell 250/500/1000/2000 IE des humanen Blutgerinnungsfaktors VIII (rDNA), Simoctocog alfa. Nach Auflösen enthält Nuwiq pro ml ungefähr 100/200/400/800 IE humanen Blutgerinnungsfaktor VIII (rDNA), Simoctocog alfa.
Pulver: weisses bis cremefarbenes, krümeliges Pulver.
Lösung: Wasser für Injektionszwecke; eine klare, farblose Flüssigkeit.
Die Aktivität (IE) wird unter Verwendung des chromogenen Tests gemäss dem Europäischen Arzneibuch bestimmt. Die spezifische Aktivität von Nuwiq beträgt ungefähr 9500 IE/mg Protein.
Indikationen/Anwendungsmöglichkeiten
Behandlung und Prophylaxe von Blutungen bei Patienten mit Hämophilie A (angeborener Faktor VIII-Mangel).
Nuwiq enthält keine pharmakologisch wirksamen Mengen des von-Willebrand-Faktors und ist daher nicht zur Behandlung des von Willebrand-Jürgens-Syndroms geeignet.
Dosierung/Anwendung
Die Behandlung muss unter der Aufsicht eines in der Behandlung der Hämophilie erfahrenen Arztes erfolgen.
Übliche Dosierung
Die Dosis und Dauer der Substitutionstherapie richtet sich nach dem Schweregrad des Faktor VIII-Mangels, dem Ort und Ausmass der Blutung und dem klinischen Zustand des Patienten.
Die Anzahl der verabreichten Faktor VIII-Einheiten wird in Internationalen Einheiten (IE) angegeben, bezogen auf den aktuellen WHO-Standard für Faktor VIII-Produkte. Die Faktor VIII-Aktivität im Plasma wird entweder in Prozent (bezogen auf humanes Normalplasma) oder in Internationalen Einheiten (bezogen auf einen internationalen Faktor VIII-Standard im Plasma) angegeben. Sowohl der einstufige Gerinnungstest als auch der chromogene Test eignen sich für die Messung der FVIII-Aktivität im Plasma. Für die klinische Anwendung und Aktivitätsvergleich mit anderen rekombinanten und plasmatischen FVIII Präparaten ist die Angabe des einstufigen Gerinnungstests üblich.
Eine Internationale Einheit (IE) Faktor VIII-Aktivität entspricht der Menge an Faktor VIII in einem Milliliter humanem Normalplasma.
Bedarfstherapie
Die Berechnung der erforderlichen Faktor VIII-Dosis basiert auf dem empirischen Ergebnis, dass eine Internationale Einheit (IE) Faktor VIII pro kg Körpergewicht die Faktor VIII-Aktivität im Plasma im Durchschnitt um 2% der normalen Aktivität oder 2 IE/dl erhöht. Die erforderliche Dosis wird anhand der folgenden Formel ermittelt:
- Erforderliche Einheiten = Körpergewicht (kg) x erwünschter Faktor VIII-Anstieg (%) (IE/dl) x 0,5 (IE/kg pro IE/dl)
- Erwarteter Faktor VIII-Anstieg (% des Normalwerts) = 2 x verabreichte IE : Körpergewicht (kg)
Die zu verabreichende Menge und die Häufigkeit der Verabreichung sollten sich immer nach der klinischen Wirksamkeit im Einzelfall richten.
Im Falle der folgenden Blutungsereignisse sollte die Faktor VIII-Aktivität nicht unter die angegebene Plasmaaktivität (in % des Normalwerts oder IE/dl) im entsprechenden Zeitraum fallen. Die Angaben in der nachstehenden Tabelle können als Dosierungsrichtwerte bei Blutungsepisoden und chirurgischen Eingriffen verwendet werden:
Schweregrad der Blutung/ | Erforderlicher Faktor VIII-Spiegel (%) (IE/dl) | Häufigkeit der Anwendung (Stunden)/Dauer der Therapie (Tage) |
|---|---|---|
Blutung | ||
Beginnende Gelenkblutungen, Muskelblutungen oder Blutungen im Mundbereich | 20–40 | Alle 12 bis 24 Stunden wiederholen. Mindestens 1 Tag, bis die durch Schmerzen erkennbare Blutung gestillt ist oder eine Heilung erreicht ist. |
Grössere Gelenkblutungen, Muskelblutung oder Hämatome | 30–60 | Infusion alle 12 bis 24 Stunden wiederholen, über 3 bis 4 Tage oder länger wiederholen, bis die Schmerzen und die akuten Beeinträchtigungen aufhören. |
Lebensbedrohliche Blutungen | 60–100 | Infusion alle 8 bis 24 Stunden wiederholen, bis der Patient ausser Gefahr ist. |
Chirurgischer Eingriff | ||
Kleinere Eingriffe | 30–60 | Alle 24 h, mindestens 1 Tag, bis eine Heilung eintritt. |
Grössere Eingriffe | 80–100 (prä- und postoperativ) | Infusion alle 8-24 h wiederholen, bis eine angemessene Wundheilung erzielt ist. Dann die Therapie für mindestens 7 Tage weiterführen, um eine Faktor-VIII-Aktivität von 30% bis 60% (IE/dl) aufrechtzuerhalten. |
Prophylaxe
Übliche Dosen zur Langzeitprophylaxe von Blutungen bei Patienten mit schwerer Hämophilie A sind 20 bis 40 IE Faktor VIII pro kg Körpergewicht in Abständen von 2 bis 3 Tagen. In manchen Fällen, insbesondere bei jüngeren Patienten, können kürzere Dosierungsintervalle oder höhere Dosen erforderlich sein.
Im Laufe der Behandlung ist es ratsam, eine angemessene Kontrolle des Faktor VIII-Spiegels durchzuführen, um die zu verabreichende Dosis und die Häufigkeit der wiederholten Infusionen entsprechend festzulegen. Vor allem bei grossen chirurgischen Eingriffen ist eine genaue Kontrolle der Substitutionstherapie mit Hilfe der Gerinnungsanalyse (Faktor VIII-Aktivität im Plasma) unerlässlich. Die Antwort auf die Gabe von Faktor VIII, gemessen als In-Vivo-Recovery und die Halbwertszeit können von Patient zu Patient variieren.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Ältere Patienten
Es liegen nur wenige Erfahrungen mit älteren Patienten vor.
Kinder und Jugendliche
Zur langfristigen Vorbeugung von Blutungen bei unter 12 jährigen Patienten wird eine Dosierung von 30 – 50 IE/kg FVIII alle 2 Tage oder dreimal wöchentlich empfohlen.
Zuvor unbehandelte Patienten
Die Sicherheit und Wirksamkeit von Nuwiq bei zuvor unbehandelten Patienten wurde in einer prospektiven klinischen Studie untersucht.
Verabreichungsschema
Es wird empfohlen, nicht mehr als 4 ml pro Minute zu verabreichen.
Art der Anwendung
Zur intravenösen Anwendung.
Hinweise zur Rekonstitution des Arzneimittels vor der Anwendung, siehe Abschnitt «Sonstige Hinweise – Hinweise für die Handhabung».
Kontraindikationen
Überempfindlichkeit gegen den Wirkstoff oder den Hilfsstoffen gemäss Zusammensetzung.
Warnhinweise und Vorsichtsmassnahmen
Überempfindlichkeit
Wie bei jedem intravenösen Proteinprodukt sind allergische Überempfindlichkeitsreaktionen möglich. Nuwiq enthält neben Faktor VIII Spuren anderer menschlicher Proteine aus den Wirtszellen. Wenn Überempfindlichkeitssymptome auftreten, sollten die Patienten angewiesen werden, das Arzneimittel sofort abzusetzen und ihren Arzt zu kontaktieren. Patienten sollten über Frühzeichen von Überempfindlichkeitsreaktionen aufgeklärt werden, wie zum Beispiel Nesselausschlag, generalisierte Urtikaria, Engegefühl in der Brust, Keuchen, Hypotonie und anaphylaktischer Schock.
Im Falle eines Schocks sind die medizinischen Standards für die Behandlung von Schockzuständen zu befolgen.
Inhibitoren
Die Bildung neutralisierender Antikörper (Inhibitoren) gegen Faktor VIII ist eine bekannte Komplikation bei der Behandlung von Hämophilie-A-Patienten. Diese Antikörper sind normalerweise IgG-Immunglobuline, die gegen die prokoagulatorische Aktivität des Faktors VIII gerichtet sind. Die Messung erfolgt mit Hilfe des modifizierten Bethesda-Tests und wird in Bethesda-Einheiten (B.E.) pro Milliliter Plasma ausgedrückt. Das Risiko, Antikörper zu entwickeln, korreliert mit dem Ausmass der Exposition gegenüber dem Faktor VIII, wobei das Risiko innerhalb der ersten 20 Expositionstage (EDs) am grössten ist. In seltenen Fällen können sich Antikörper nach den ersten 100 EDs bilden.
Bei vorbehandelten Patienten mit mehr als 100 EDs, die in der Vergangenheit bereits Antikörper entwickelt hatten, wurden beim Wechsel von einem Faktor VIII-Produkt zu einem anderen Fälle wiederkehrender (niedrigtitriger) Antikörper beobachtet. Daher wird empfohlen, Patienten nach jedem Produktwechsel sorgfältig auf das Auftreten eines Antikörpers hin zu überwachen.
Die Immunogenität von Nuwiq wurde bei zuvor unbehandelten Patienten mit schwerer Hämophilie A (<1% FVIII:C) in einer prospektiven offenen klinischen Studie untersucht. Von 105 Patienten, die Nuwiq erhielten und nach Beginn der Behandlung mindestens einmal auf neutralisierende Antikörper überprüft wurden, entwickelten 17 Patienten (16,2%) neutralisierende Antikörper mit hohem Titer und 11 Patienten (10,5%) neutralisierende Antikörper mit niedrigem Titer (5 davon waren transiente Antikörper, die durch Fortsetzung der prophylaktischen Behandlung ohne Änderung der Dosierung eliminiert wurden). Von den 28 Patienten, die einen Antikörper entwickelten, hatten 25 weniger als 20 EDs vor dem Nachweis. Die mittleren Zeiten bis zur Entwicklung von Antikörper mit hohem Titer bzw. niedrigem Titer betrugen 9,0 EDs (Bereich 4–24 EDs) bzw. 12,0 EDs (Bereich 6–34 EDs). Bei Patienten mit Non-Null F8 Gen Mutationen wurden keine neutralisierenden Antikörper nachgewiesen.
Im Allgemeinen sollten alle Patienten, die mit Blutgerinnungsfaktor VIII behandelt werden, durch sorgfältige klinische Beobachtung und Labortests auf die Entwicklung eines Antikörpers hin überwacht werden. Wenn die erwarteten Faktor VIII-Spiegel im Plasma nicht erreicht werden oder die Blutung nicht durch eine angemessene Dosis unter Kontrolle gebracht werden kann, sollte auf das Vorhandensein von Faktor VIII-Antikörper getestet werden. Bei Patienten mit einem hohen Antikörperspiegel ist eine Faktor VIII-Therapie möglicherweise nicht wirksam und es sollten andere Behandlungsoptionen, wie z.B. Immuntoleranzinduktion (ITI) in Betracht gezogen werden. Die Behandlung solcher Patienten sollte von Ärzten geleitet werden, die im Umgang mit Hämophilie und Faktor VIII-Antikörper Erfahrung haben.
Katheter-assoziierte Komplikationen
Wenn ein zentraler Venenkatheter (ZVK) erforderlich ist, sollte das Risiko ZVK-assoziierter Komplikationen, einschliesslich lokaler Infektionen, Bakteriämie und Thrombose an der Katheterstelle berücksichtigt werden.
Es wird dringend empfohlen, bei jeder Verabreichung von Nuwiq den Namen und die Chargennummer des Produkts zu dokumentieren, damit jederzeit ein Zusammenhang zwischen Patient und der Produktcharge hergestellt werden kann.
Kinder und Jugendliche
Die aufgelisteten Warnungen und Vorsichtsmassnahmen gelten für Erwachsene und Kinder gleichermassen.
Hinweis zu den sonstigen Bestandteilen (Natriumgehalt)
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Durchstechflasche, d.h. es ist nahezu «natrium-frei».
Allerdings besteht die Möglichkeit, dass ein Patient, je nach Körpergewicht und Dosierung, mehr als eine Durchstechflasche erhält (siehe Rubrik «Darreichungsform und Wirkstoffmenge pro Einheit» für den Gehalt pro Durchstechflasche). Dies muss von Patienten, die eine natriumkontrollierte Diät erhalten, berücksichtigt werden.
Interaktionen
Es wurden keine Interaktionsstudien zu Wechselwirkungen von Nuwiq durchgeführt.
Schwangerschaft/Stillzeit
Schwangerschaft
Es gibt keine hinreichenden Daten zur Anwendung bei Schwangeren.
Es liegen keine hinreichenden tierexperimentellen Studien zur Auswirkung auf Schwangerschaft, Embryonalentwicklung, Entwicklung des Föten und/oder die postnatale Entwicklung vor. Das potentielle Risiko für den Menschen ist nicht bekannt.
Aufgrund des seltenen Auftretens von Hämophilie A bei Frauen liegen keine Erfahrungen über die Anwendung von Nuwiq bei Schwangeren vor. Daher sollte Nuwiq während der Schwangerschaft nur dann angewandt werden, wenn dies unbedingt erforderlich ist.
Stillzeit
Aufgrund des seltenen Auftretens von Hämophilie A bei Frauen liegen keine Erfahrungen über die Anwendung von Nuwiq bei stillenden Müttern vor. Daher sollte Nuwiq während der Stillzeit nur dann angewandt werden, wenn dies unbedingt erforderlich ist.
Fertilität
Es liegen keine Daten zur Beeinflussung der Fertilität vor.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Nuwiq hat keinen Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen.
Unerwünschte Wirkungen
Zusammenfassung des Sicherheitsprofils
In seltenen Fällen wurden bei Faktor VIII-Präparaten Überempfindlichkeits- oder allergische Reaktionen (wie z.B. Angioödem, Brennen und Stechen an der Infusionsstelle, Schüttelfrost, Gesichtsrötung und Hitzegefühl (Flushing), generalisierte Urtikaria, Kopfschmerzen, Nesselausschlag, Hypotonie, Lethargie, Übelkeit, Unruhe, Tachykardie, Engegefühl in der Brust, Kribbeln, Erbrechen, Keuchen) beobachtet. In einigen Fällen können sich diese Symptome/Reaktionen zur schweren Anaphylaxie (einschliesslich Schock) entwickeln.
Patienten mit Hämophilie A können neutralisierende Antikörper (Inhibitoren) gegen Faktor VIII entwickeln. Falls derartige Antikörper auftreten, äussert sich dies in Form einer unzureichenden klinischen Wirksamkeit. In solchen Fällen wird die Kontaktaufnahme mit einem spezialisierten Hämophiliezentrum empfohlen.
Tabellarische Auflistung von Nebenwirkungen
In klinischen Studien mit Nuwiq an zuvor behandelten Kindern (2 bis 11 Jahre, n = 58), Jugendlichen (12 bis 17 Jahre, n = 3) und erwachsenen Patienten (n = 129) mit schwerer Hämophilie A wurden bei 8 Patienten (4 Erwachsenen, 4 Kindern) insgesamt 12 unerwünschte Arzneimittelwirkungen (UAWs) (8 bei Erwachsenen, 4 bei Kindern) berichtet. In einer klinischen Studie mit 108 vorher unbehandelten Patienten (im Alter zwischen 0-146 Monaten) mit schwerer Hämophilie A wurden folgende UAWs berichtet: neutralisierende Antikörper gegen Faktor VIII (Inhibitoren) bei 28 Patienten, Pyrexie bei 20 Patienten, Ausschlag bei 5 Patienten, Überempfindlichkeit bei 2 Patienten, und Schüttelfrost und Urtikaria (jeweils bei einem Patienten).
Die nachfolgende Tabelle 1 entspricht der MedDRA-Systemorganklassifizierung (SOC und Preferred Term-Level).
Die Häufigkeiten wurden gemäss folgender Konvention beurteilt: sehr häufig (≥1/10); häufig (≥1/100 bis <1/10); gelegentlich (≥1/1'000 bis <1/100); selten (≥1/10'000 bis <1/1'000); sehr selten (<1/10'000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Innerhalb jeder Häufigkeitsgruppe sind die unerwünschten Wirkungen nach abnehmendem Schweregrad angegeben.
Tabelle 1. Häufigkeit des Auftretens von unerwünschten Arzneimittelwirkungen (UAWs) pro Patient in klinischen Studien bei 298 Patienten mit schwerer Hämophilie A
MedDRA-Systemorganklasse | Unerwünschte Wirkungen | Häufigkeit |
Blut- und Lymphsystem Störungen | Antikörper gegen Faktor VIII | gelegentlich (bei zuvor behandelten Patienten)# sehr häufig (bei nicht vorbehandelten Patienten)# |
Erkrankungen des Immunsystems | Überempfindlichkeit | gelegentlich* |
Erkrankungen des Nervensystems | Parästhesien; Kopfschmerzen | gelegentlich* |
Erkrankungen des Ohrs und des Labyrinths | Schwindel | gelegentlich* |
Erkrankungen des Gastrointestinaltrakts | Mundtrockenheit | gelegentlich* |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | Rückenschmerzen | gelegentlich* |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | Pyrexie Entzündung an der Injektionsstelle; Schmerzen an der Injektionsstelle; Schüttelfrost Unwohlsein | häufig* gelegentlich* |
Untersuchungen | Nicht-neutralisierende Antikörper gegen Faktor VIII (bei zuvor behandelten Patienten) | gelegentlich* |
Atemwege, Brustkorb und mediastinale Störungen | Dyspnoe | gelegentlich* |
Haut- und Unterhauterkrankungen | Ausschlag Urtikaria | häufig* gelegentlich* |
* Berechnet als Patienten mit UAWs pro Gesamtzahl von 298 Studienpatienten, davon 190 zuvor behandelte Patienten und 108 nicht vorbehandelten Patienten.
# Die Häufigkeit basiert auf Studien mit allen FVIII-Produkten, die Patienten mit schwerer Hämophilie A einschlossen.
Beschreibung ausgewählter Nebenwirkungen
Ein nicht-neutralisierender Antikörper gegen Faktor VIII wurde bei einem erwachsenen Patienten festgestellt (siehe Tabelle 1). Die Probe wurde vom Zentrallabor in 8 Verdünnungen getestet. Nur bei Verdünnungsfaktor 1 war das Ergebnis positiv und der Antikörpertiter war sehr niedrig. Eine inhibitorische Aktivität, gemäss modifiziertem Bethesda-Test, wurde bei diesem Patienten nicht festgestellt. Die klinische Wirksamkeit und die In-vivo-Recovery von Nuwiq waren bei diesem Patienten nicht beeinträchtigt.
Kinder und Jugendliche
Es wird davon ausgegangen, dass die Häufigkeit, die Art und der Schweregrad von Nebenwirkungen bei Kindern die gleichen sind, wie bei Erwachsenen.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Es wurden keine Fälle von Überdosierung berichtet.
Eigenschaften/Wirkungen
ATC-Code
B02BD02
Wirkungsmechanismus
Simoctocog alfa [humaner Blutgerinnungsfaktor VIII (rDNA)] ist ein gereinigtes Protein, bestehend aus 1440 Aminosäuren. Die Aminosäuresequenz ist vergleichbar mit der 90- und 80-kDa-Form des humanen Plasma-Faktor-VIII (d.h., die B-Domäne ist entfernt). Nuwiq wird mittels rekombinanter DNS-Technologie in genetisch veränderten, menschlichen, embryonalen Nierenzellen (HEK) der Zelllinie HEK-293F hergestellt. Materialien menschlichen oder tierischen Ursprunges werden weder während der Herstellung noch dem fertigen Produkt hinzugefügt.
Hämophilie A ist eine X chromosomalgebundene, erbliche Störung der Blutgerinnung, aufgrund erniedrigter Faktor VIII:C -Spiegel. Als Folge treten starke Blutungen in Gelenken, Muskeln oder inneren Organen auf. Diese können spontan oder als Folge von Unfällen oder chirurgischen Eingriffen entstehen. Die Substitutionstherapie hebt den Plasmaspiegel des Faktor VIII an und ermöglicht so eine vorübergehende Korrektur des Faktor VIII-Mangels und der Blutungsneigung.
Pharmakodynamik
Der Faktor VIII/von-Willebrand-Faktor-Komplex besteht aus zwei Molekülen (Faktor VIII und von-Willebrand-Faktor) mit verschiedenen physiologischen Funktionen. Wird einem Hämophilie-A-Patienten Faktor VIII injiziert, so bindet dieser im Blutkreislauf an den von-Willebrand-Faktor. Aktivierter Faktor VIII wirkt als Kofaktor für aktivierten Faktor IX und beschleunigt die Umwandlung von Faktor X in aktivierten Faktor X. Der aktivierte Faktor X wandelt Prothrombin in Thrombin um. Thrombin wandelt dann Fibrinogen in Fibrin um und führt so zur Bildung eines Gerinnsels.
Klinische Wirksamkeit
Die Immunogenität von Nuwiq wurde in klinischen Studien bei 135 zuvor behandelten Patienten mit schwerer Hämophilie A untersucht (74 Erwachsene sowie 61 Kinder). In 4 Patienten konnten nicht-neutralisierende Faktor VIII Antikörper nachgewiesen werden. 3 von den 4 Patienten hatten bereits einen nicht-neutralisierenden Antikörper vor der Verabreichung von Nuwiq. Ein nicht-neutralisierender Antikörper gegen Faktor VIII wurde bei einem erwachsenen Patienten festgestellt. Die Probe wurde vom Zentrallabor in 8 Verdünnungen getestet. Nur bei Verdünnungsfaktor 1 war das Ergebnis positiv und der Antikörpertiter war sehr niedrig. Eine inhibitorische Aktivität, gemäss modifiziertem Bethesda-Test, wurde bei diesem Patienten nicht festgestellt. Die klinische Wirksamkeit und die In-vivo-Recovery von Nuwiq waren bei diesem Patienten nicht beeinträchtigt.
In einer klinischen Studie mit 32 erwachsenen Patienten mit schwerer Hämophilie A lag der mittlere Verbrauch von Nuwiq zur Prophylaxe bei 468,7 IE/kg/Monat. Die mittlere Dosis zur Behandlung von Episoden mit Durchbruchsblutungen betrug 33,0 IE/kg bei diesen Patienten unter prophylaktischer Behandlung. In einer anderen klinischen Studie erhielten 22 erwachsene Patienten eine Bedarfsbehandlung. Insgesamt wurden 986 Blutungsepisoden mit einer mittleren Dosis von 30,9 IE/kg behandelt. Im Allgemeinen waren für leichte Blutungen etwas niedrigere und für schwerere Blutungen bis zu dreifach höhere mittlere Dosen erforderlich.
Individualisierte Prophylaxe: Die individualisierte PK-basierte Prophylaxe wurde in 66 erwachsenen PTPs mit schwerer Hämophilie A untersucht. Nach einer 1-3-monatigen Standard-Prophylaxe-Phase (jeden zweiten Tag oder 3-mal wöchentliche Dosierung), konnten 44 (67%) Patienten auf der Grundlage ihrer PK-Bewertung auf ein Dosierschema umgestellt werden, und 40 absolvierten die 6 Monate Prophylaxe nach dem empfohlenen kalkulierten Dosierungs- und Behandlungsschema. Von diesen Patienten wurden 34 (85%) zweimal wöchentlich oder seltener behandelt. Bei 33 (82,5%) Patienten traten keine Blutungen auf, und 36 (90,0%) Patienten hatten keine spontanen Blutungen. Die annualisierte Blutungsrate (ABR; Mittelwert ± SD) betrug 1,2 ± 3,9 und die mittlere Dosis ± SD lag bei 52,2 ± 12,2 I.E./kg pro Injektion bzw. 99,7 ± 25,6 I.E./kg pro Woche.
Es gilt zu beachten, dass die ABR zwischen verschiedenen Faktorkonzentraten und zwischen verschiedenen klinischen Studien nicht vergleichbar ist.
Sicherheit und Wirksamkeit bei pädiatrischen Patienten
Die Daten wurden bei 29 zuvor behandelten Kindern zwischen 2 und 5 Jahren, 31 Kindern zwischen 6 und 12 Jahren und einem Jugendlichen von 14 Jahren erhoben. Die mittlere Dosis pro prophylaktischer Infusion lag bei 37,8 IE/kg. Zwanzig Patienten verwendeten mittlere Dosen von mehr als 45 IE/kg. Der mittlere Verbrauch von Nuwiq zur Prophylaxe pro Monat lag bei 521,9 IE/kg. Für die Behandlung von Blutungen bei Kindern war eine höhere Dosis Nuwiq (43,9 IE/kg) erforderlich als bei Erwachsenen (33,0 IE/kg) und eine höhere mittlere Dosis war für die Behandlung von moderaten bis schweren als für leichte Blutungen (78,2 IE/kg vs. 41,7 IE/kg) erforderlich. Bei jüngeren Kindern waren im Allgemeinen höhere mittlere Dosen erforderlich (6-12 Jahre: 43,9 IE/kg; 2-5 Jahre: 52,6 IE/kg). Diese Daten wurden durch die Langzeitbeobachtung von 49 dieser Kinder, die für einen zusätzlichen Medianzeitraum von ca. 30 Monaten behandelt wurden (Bereich von 9,5 bis 52 Monaten), bestätigt; in diesem Zeitraum hatten 45% der Kinder keine spontanen Blutungen.
Pharmakokinetik
Absorption
Nicht zutreffend.
Distribution
FVIII verteilt sich im Plasma.
Metabolismus
Nicht zutreffend.
Elimination
Siehe Tabellen 2 – 4.
Tabelle 2. Pharmakokinetik-(PK)-Parameter für Nuwiq (Dosis: 50 IE/kg) bei erwachsenen vorbehandelten Patienten (im Alter von 18-65 Jahren) mit schwerer Hämophilie A (n = 20)
PK-Parameter | Chromogener Test | Einstufiger Gerinnungstest |
|---|---|---|
Mittelwert ± SD | Mittelwert ± SD | |
AUCnorm (h*IE/ml/(IE/kg)) | 0,39 ± 0,14 | 0,37 ± 0,11 |
T½ (h) | 14,7 ± 10,4 | 17,0 ± 11,8 |
IVR (%/IE/kg) | 2,5 ± 0,4 | 2,2 ± 0,3 |
CL (ml/h/kg) | 3,0 ± 1,2 | 2,9 ± 1,0 |
AUC = Fläche unter der Kurve (FVIII:C), T½ = terminale Halbwertszeit,
IVR = inkrementelle In-Vivo-Recovery, CL = Clearance, SD = Standardabweichung
Kinetik spezieller Patientengruppen
Gewichtsangepasste Untergruppen
Bei den in der PK Studie eingeschlossenen erwachsenen präadipösen (BMI 25-30 kg/m2) und adipösen Patienten (BMI >30 kg/m2) war die Clearance höher als bei den Normalgewichtigen.
Kinder und Jugendliche
Tabelle 3. PK-Parameter für Nuwiq (Dosis: 50 IE/kg) bei vorbehandelten Kindern im Alter von 6 bis 12 Jahren mit schwerer Hämophilie A (n = 12)
PK-Parameter | Chromogener Test | Einstufiger Gerinnungstest |
|---|---|---|
Mittelwert ± SD | Mittelwert ± SD | |
AUCnorm (h*IE/ml/(IE/kg)) | 0,25 ± 0,1 | 0,26 ± 0,1 |
T½(h) | 10,0 ± 1,9 | 13,1 ± 2,6 |
IVR (%/IE/kg) | 1,9 ± 0,4 | 1,6 ± 0,4 |
CL (ml/h/kg) | 4,3 ± 1,2 | 4,1 ± 0,9 |
AUC = Fläche unter der Kurve (FVIII:C), T½ = terminale Halbwertszeit,
IVR = inkrementelle In-Vivo-Recovery, CL = Clearance, SD = Standardabweichung
Tabelle 4. PK-Parameter für Nuwiq (Dosis: 50 IE/kg) bei vorbehandelten Kindern im Alter von 2 bis 5 Jahren mit schwerer Hämophilie A (n = 13)
PK-Parameter | Chromogener Test | Einstufiger Gerinnungstest |
|---|---|---|
Mittelwert ± SD | Mittelwert ± SD | |
AUCnorm (h*IE/ml/(IE/kg)) | 0,22 ± 0,1 | 0,22 ± 0,1 |
T½(h) | 9,5 ± 3,3 | 11,9 ± 5,4 |
IVR (%/IE/kg) | 1,9 ± 0,3 | 1,6 ± 0,2 |
CL (ml/h/kg) | 5,4 ± 2,4 | 5,4 ±2,3 |
AUC = Fläche unter der Kurve (FVIII:C), T½ = terminale Halbwertszeit,
IVR = inkrementelle In-Vivo-Recovery, CL = Clearance, SD = Standardabweichung
Wie aus der Literatur bekannt, waren bei jüngeren Kindern Recovery und Halbwertszeit niedriger und die Clearance höher als bei Erwachsenen, was teilweise am bekanntermassen höheren Plasmavolumen pro Kilogramm Körpergewicht bei jüngeren Patienten liegen könnte.
Präklinische Daten
Toxikologische Studien zeigten, dass die lokale intravenöse Verabreichung und systemische Exposition von Labortieren (Ratten und Cynomolgus-Affen) gut vertragen wurde. Die Bedeutung der nach wiederholter Simoctocog alfa-Gabe in Affen beobachteten erniedrigten Thymusgewichte ist unklar.
Aufgrund der Immunreaktion auf heterologe Proteine bei allen nicht-humanen Säugetierarten wurden mit Nuwiq keine spezifischen Studien mit wiederholter Verabreichung über einen längeren Zeitraum durchgeführt (wie z.B. Studien zu Reproduktionstoxizität, chronischer Toxizität und Kanzerogenität).
Es wurden keine Studien zum mutagenen Potential von Nuwiq durchgeführt.
Ex-Vivo-Untersuchungen mit Hilfe eines kommerziellen Test-Kits zur Quantifizierung der T-Zell-Antwort auf Proteintherapeutika zeigen eine zu vergleichbaren Präparaten mindestens gleichermassen ausgeprägte Immunogenität an.
Sonstige Hinweise
Inkompatibilitäten
Da keine Verträglichkeitsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Es dürfen nur die mitgelieferten Infusionssets verwendet werden, da Therapieversagen als Folge einer Adsorption von humanem Gerinnungsfaktor VIII an der inneren Oberfläche mancher Infusionssets auftreten kann.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «Verw. bis/EXP» bezeichneten Datum verwendet werden.
Während der Haltbarkeitsfrist kann das Präparat für einen Zeitraum von nicht mehr als 1 Monat bei Raumtemperatur (bis zu 25 °C) aufbewahrt werden. Sobald das Präparat aus dem Kühlschrank genommen wird, darf es nicht wieder in diesen zurückgelegt werden. Vermerken Sie bitte den Beginn der Aufbewahrung bei Raumtemperatur auf der Packung. Die Durchstechflasche muss in der Originalverpackung zum Schutz vor Licht gelagert werden.
Nach der Rekonstitution
Aus mikrobiologischer Sicht sollte das gebrauchsfertige Arzneimittel sofort nach Rekonstitution verwendet werden. Geschieht das nicht, so ist das rekonstituierte Präparat bei einer Lagertemperatur von ≤25 °C innerhalb von 3 Stunden zu verwenden. Das rekonstituierte Präparat muss in der Durchstechflasche aufbewahrt werden.
Nicht verwendete Präparate, die länger als 3 Stunden bei Raumtemperatur gelagert wurden, müssen entsorgt werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8 °C) lagern.
Nicht einfrieren. In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Zu den Lagerungsbedingungen für das rekonstituierte Präparat siehe unter «Sonstige Hinweise: Haltbarkeit – Nach der Rekonstitution».
Entsorgung
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Hinweise für die Handhabung
Das Pulver darf nur mit dem mitgelieferten Lösungsmittel (2,5 ml Wasser für Injektionszwecke) und unter Verwendung des mitgelieferten Injektionssets rekonstituiert werden. Die Durchstechflasche sollte vorsichtig geschwenkt werden, bis das gesamte Pulver aufgelöst ist. Nach der Rekonstitution sollte die Lösung wieder in die Spritze aufgezogen werden.
Das rekonstituierte Arzneimittel muss vor der Verabreichung per Sichtkontrolle auf Partikel und Verfärbungen hin geprüft werden. Das rekonstituierte Arzneimittel ist eine klare, farblose Lösung, die frei von Fremdkörpern ist und einen pH-Wert zwischen 6,5 und 7,5 aufweist. Verwenden Sie keine Lösungen, die trüb sind oder Ablagerungen enthalten.
Rekonstitution:
- Bringen Sie die Lösungsmittelspritze (mit Wasser für Injektionszwecke) und das Pulver in der verschlossenen Durchstechflasche vor der Anwendung auf Zimmertemperatur. Sie können dazu die Behälter in den Händen halten, bis sie sich so warm wie Ihre Hände anfühlen. Wenden Sie keine andere Methode zum Aufwärmen der Durchstechflasche und der Fertigspritze an. Diese Temperatur muss während der Rekonstitution beibehalten werden.
- Entfernen Sie die Flipp-off-Kappe aus Plastik von der Durchstechflasche mit dem Pulver, so dass der mittlere Teil des Gummistopfens sichtbar wird. Entfernen Sie nicht den grauen Stopfen oder den Metallring (Bördelkappe) am oberen Ende der Durchstechflasche.

- Wischen Sie den Gummistopfen der Durchstechflasche mit einem Alkoholtupfer ab. Lassen Sie den Alkohol trocknen.
- Ziehen Sie die Schutzfolie von der Verpackung des Durchstechflaschen-Adapters ab. Nehmen Sie den Adapter nicht aus seiner Verpackung.

- Stellen Sie die Durchstechflasche mit dem Pulver auf eine ebene Unterlage und halten Sie sie fest. Nehmen Sie den Adapter mit seiner Verpackung und platzieren Sie den Durchstechflaschen-Adapter mittig über dem Gummistopfen der Durchstechflasche mit dem Pulver. Drücken Sie die Adapterverpackung mit dem Adapter kräftig auf, bis der Adapterdorn den Gummistopfen durchdringt. Dabei rastet der Adapter auf der Durchstechflasche ein.
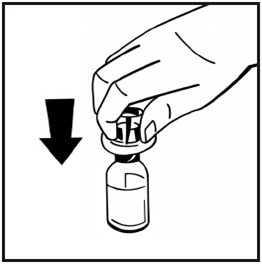
- Ziehen Sie die Schutzfolie von der Verpackung der Fertigspritze ab. Halten Sie den Spritzenstempel am Ende und berühren Sie nicht den Schaft. Stecken Sie das Ende des Spritzenstempels mit dem Gewinde auf den Kolben der Lösungsmittelspritze. Drehen Sie den Stempel im Uhrzeigersinn, bis Sie einen leichten Widerstand spüren.

- Brechen Sie die Spritzenkappe aus Plastik von der Lösungsmittelspritze an der Perforation ab. Berühren Sie nicht die Innenseite der Kappe oder der Spritzenspitze. Wird die Lösung nicht sofort verwendet, verschliessen Sie die gefüllte Spritze mit der Spritzenkappe zur Aufbewahrung.

- Entfernen Sie die Adapterverpackung und entsorgen Sie diese.
- Verbinden Sie die Lösungsmittelspritze fest mit dem Durchstechflaschen-Adapter, indem Sie sie im Uhrzeigersinn drehen, bis ein Widerstand spürbar wird.

- Injizieren Sie langsam das gesamte Lösungsmittel in die Durchstechflasche mit dem Pulver, indem Sie den Spritzenstempel nach unten drücken.
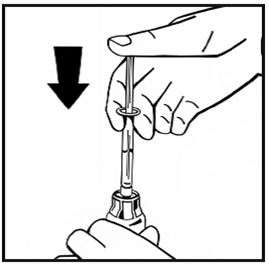
- Zum Auflösen des Pulvers schwenken Sie die Durchstechflasche einige Male leicht im Kreis, ohne dabei die Spritze zu entfernen. Nicht schütteln. Warten Sie bis sich das Pulver vollständig aufgelöst hat.
- Prüfen Sie die fertige Lösung vor der Verabreichung visuell auf Partikel. Die Lösung sollte klar und farblos sein und frei von sichtbaren Partikeln. Verwenden Sie keine Lösungen, die trüb sind oder Ablagerungen enthalten.
- Drehen Sie die mit der Spritze verbundene Durchstechflasche auf den Kopf und ziehen Sie die fertige Lösung langsam in die Spritze auf. Stellen Sie sicher, dass der gesamte Inhalt der Durchstechflasche in die Spritze überführt wird.

- Ziehen Sie die gefüllte Spritze mit einer Drehbewegung gegen den Uhrzeigersinn aus dem Durchstechflaschen-Adapter und verwerfen Sie die leere Durchstechflasche.
- Die Lösung steht nun zur sofortigen Verwendung bereit. Nicht im Kühlschrank lagern.
- Reinigen Sie die vorgesehene Injektionsstelle mit einem der mitgelieferten Alkoholtupfer.
- Verbinden Sie das mitgelieferte Infusionsset mit der Spritze. Führen Sie die Nadel der Flügelkanüle in die ausgewählte Vene ein. Wenn Sie die Vene vor der Punktion gestaut haben, damit Sie sie besser sehen können, müssen Sie die Stauung öffnen, bevor Sie mit der Injektion der Lösung beginnen. Es darf kein Blut in die Spritze gelangen, da dies zur Bildung von Blutgerinnseln führen könnte.
- Injizieren Sie die Lösung langsam in die Vene. Die Injektionsgeschwindigkeit sollte höchstens 4 ml pro Minute betragen.
Wenn Sie mehr als eine Durchstechflasche mit Pulver für eine Behandlung benötigen, können Sie dieselbe Flügelkanüle verwenden. Der Durchstechflaschen-Adapter und die Spritze sind nur für den einmaligen Gebrauch bestimmt.
Zulassungsnummer
65551 (Swissmedic).
Zulassungsinhaberin
Octapharma AG, 8853 Lachen.
Stand der Information
September 2020.
Composizione
Principi attivi
Simoctocog alfa (fattore VIII della coagulazione ricombinante di natura umana).
Sostanze ausiliarie
Saccarosio, sodio cloruro, calcio cloruro biidrato, arginina cloridrato, sodio citrato biidrato, poloxamer 188 (corrisponde al contenuto di sodio totale di 18,4 mg/fiala).
Solvente: acqua per preparazioni iniettabili.
Forma farmaceutica e quantità di principio attivo per unità
Polvere e solvente per soluzione iniettabile.
Ogni flaconcino contiene nominalmente 250/500/1000/2000 UI di fattore VIII della coagulazione (rDNA) di natura umana, simoctocog alfa. Nuwiq contiene circa 100/200/400/800 UI di fattore VIII della coagulazione (rDNA) di natura umana, simoctocog alfa, dopo la ricostituzione.
Polvere: polvere friabile di colore da bianco ad avorio.
Soluzione: acqua per preparazioni iniettabili; liquido trasparente e incolore.
La potenza (UI) è determinata usando il metodo cromogenico della Farmacopea europea. L'attività specifica di Nuwiq è di circa 9500 UI/mg di proteina.
Indicazioni/Possibilità d'impiego
Trattamento e profilassi degli eventi emorragici in pazienti con emofilia A (deficit congenito del fattore VIII).
Nuwiq non contiene nessuna quantità farmacologicamente efficace del fattore di von Willebrand, pertanto non è indicato per il trattamento della malattia di von Willebrand-Jürgens.
Posologia/Impiego
Il trattamento deve avvenire sotto la supervisione di un medico esperto nel trattamento dell'emofilia.
Posologia abituale
La dose e la durata della terapia sostitutiva dipendono dalla gravità del deficit di fattore VIII, dalla localizzazione ed estensione dell'emorragia e dalle condizioni cliniche del paziente.
Il numero di unità di fattore VIII somministrate è espresso in unità internazionali (UI), che sono correlate allo standard OMS vigente per i prodotti a base di fattore VIII. L'attività plasmatica del fattore VIII viene espressa in percentuale (riferita al plasma umano normale) o in unità internazionali (riferite allo standard internazionale per il fattore VIII plasmatico). Sia il saggio di coagulazione monofase che il metodo cromogenico sono adatti a misurare l'attività FVIII nel plasma. Per l'applicazione clinica e il confronto dell'attività con altri preparati FVIII ricombinanti e plasmatici, è solitamente indicato il saggio di coagulazione monofase.
Un'unità internazionale (UI) di attività di fattore VIII è equivalente alla quantità di fattore VIII contenuta in un millilitro di plasma umano normale.
Trattamento al bisogno
Il calcolo della dose necessaria di fattore VIII si basa sulla considerazione empirica che l'unità internazionale (UI) di fattore VIII per kg di peso corporeo aumenta l'attività plasmatica del fattore VIII in media del 2% dell'attività normale o di 2 UI/dl. La dose necessaria viene determinata servendosi della formula seguente:
- Unità richieste = peso corporeo (kg) x aumento di fattore VIII desiderato (%) (UI/dl) x 0,5 (UI/kg per UI/dl)
- Aumento di fattore VIII previsto (% del normale) = 2 x UI somministrate : peso corporeo (kg)
La quantità da somministrare e la frequenza di somministrazione devono essere sempre orientate all'efficacia clinica nei singoli casi.
Nel caso degli eventi emorragici seguenti, l'attività del fattore VIII non deve calare al di sotto del livello di attività plasmatica indicato (in % del normale o UI/dl) nel periodo corrispondente. La tabella qui di seguito è utilizzabile per guidare il dosaggio negli episodi emorragici e negli interventi chirurgici.
Grado dell'emorragia/ | Livello di fattore VIII necessario (%) (UI/dl) | Frequenza delle dosi (ore)/Durata della terapia (giorni) |
|---|---|---|
Emorragia | ||
Emartro in fase iniziale, emorragia muscolare o emorragia orale | 20–40 | Ripetere ogni 12-24 ore per almeno 1 giorno, finché l'episodio emorragico nella misura indicata dal dolore non si risolve o non si giunge a guarigione. |
Emartro più esteso, emorragia muscolare o ematoma | 30–60 | Ripetere l'infusione ogni 12-24 ore per 3-4 giorni o più, finché il dolore e la disabilità acuta non si risolvono. |
Emorragie potenzialmente fatali | 60–100 | Ripetere l'infusione ogni 8-24 ore finché la minaccia non scompare. |
Intervento chirurgico | ||
Intervento chirurgico minore tra cui estrazione dentaria | 30–60 | Ogni 24 ore, per almeno 1 giorno, finché non si giunge a guarigione. |
Intervento chirurgico maggiore | 80–100 (pre e postoperatorio) | Ripetere l'infusione ogni 8–24 ore finché non si raggiunge un'adeguata guarigione della ferita, quindi terapia per almeno 7 giorni per mantenere un'attività del fattore VIII del 30-60% (UI/dl). |
Profilassi
Per la profilassi antiemorragica a lungo termine in pazienti affetti da emofilia A grave, le dosi consuete sono di 20-40 UI di fattore VIII per kg di peso corporeo a intervalli di 2-3 giorni. In alcuni casi, soprattutto nei pazienti più giovani, possono essere necessari intervalli di somministrazione più brevi o dosi più elevate.
Nel corso del trattamento è consigliabile effettuare un adeguato controllo del livello del fattore VIII per determinare la dose da somministrare e la frequenza delle ripetute infusioni. In particolare, in caso di interventi chirurgici maggiori, è indispensabile effettuare un accurato monitoraggio della terapia sostitutiva mediante analisi della coagulazione (attività plasmatica del fattore VIII). La risposta alla somministrazione del fattore VIII, misurata come recupero in vivo, e l'emivita possono variare da paziente a paziente.
Per garantire la tracciabilità dei farmaci biotecnologici si raccomanda di documentare il nome e il numero di lotto del prodotto ad ogni somministrazione.
Pazienti anziani
Sono disponibili solo poche esperienze con pazienti anziani.
Bambini e adolescenti
Per la prevenzione a lungo termine delle emorragie nei pazienti di età inferiore ai 12 anni, si raccomanda un dosaggio di 30-50 UI/kg di FVIII ogni 2 giorni o tre volte alla settimana.
Pazienti non trattati in precedenza
La sicurezza e l'efficacia di Nuwiq in pazienti non trattati in precedenza è stata esaminata in uno studio clinico prospettico.
Regime posologico
Si raccomanda di non somministrare più di 4 ml al minuto.
Tipo di applicazione
Per applicazione endovenosa
Per le istruzioni sulla ricostituzione del medicinale prima della somministrazione, vedere paragrafo «Altre indicazioni – Indicazioni per la manipolazione».
Controindicazioni
Ipersensibilità al principio attivo o a una qualsiasi delle sostanze ausiliarie secondo la composizione.
Avvertenze e misure precauzionali
Ipersensibilità
Come per qualsiasi prodotto per uso endovenoso a base di proteine, sono possibili reazioni da ipersensibilità di tipo allergico. Nuwiq contiene tracce di proteine delle cellule ospiti umane diverse dal fattore VIII. Se insorgono sintomi di ipersensibilità, i pazienti devono essere avvisati di interrompere immediatamente l'uso del medicinale e contattare il proprio medico. I pazienti devono essere informati circa i primi segni delle reazioni di ipersensibilità che comprendono orticaria, orticaria generalizzata, costrizione toracica, rantolo, ipotensione e anafilassi.
In caso di shock, deve essere attuato il trattamento medico standard per lo shock.
Inibitori
La formazione di anticorpi neutralizzanti (inibitori) verso il fattore VIII rappresenta una complicanza nota nel trattamento di soggetti affetti da emofilia A. Tali anticorpi sono generalmente immunoglobuline IgG dirette contro l'attività procoagulante del fattore VIII. La quantificazione avviene per mezzo del saggio modificato Bethesda ed è espressa in unità Bethesda (UB) per millilitro di plasma. Il rischio di sviluppare anticorpi è correlato al tempo di esposizione al fattore VIII; tale rischio è maggiore entro i primi 20 giorni di esposizione. In casi rari possono formarsi anticorpi dopo i primi 100 giorni di esposizione.
Casi di comparsa di anticorpi ricorrenti (a basso titolo) sono stati osservati a seguito del passaggio da un prodotto a base di fattore VIII ad un altro, in pazienti già trattati in precedenza con più di 100 giorni di esposizione e che in passato avevano già sviluppato anticorpi. Si raccomanda pertanto di monitorare attentamente i pazienti per la ricomparsa di anticorpi dopo qualsiasi passaggio da un prodotto a un altro.
L'immunogenicità di Nuwiq è stata valutata in uno studio clinico prospettico aperto in pazienti affetti da emofilia A grave (<1% FVIII:C) e mai trattati in precedenza. Di 105 pazienti, ai quali era stato somministrato Nuwiq e che dopo l'inizio del trattamento sono stati esaminati almeno una volta per gli anticorpi neutralizzanti, in 17 pazienti (16,2%) è stato segnalato lo sviluppo di anticorpi neutralizzanti ad alto titolo e in 11 pazienti (10,5%) è stato segnalato lo sviluppo di anticorpi neutralizzanti con basso titolo (in 5 dei quali si è trattato di anticorpi transienti che sono stati eliminati proseguendo con il trattamento, senza necessità di modificare il dosaggio). Dei 28 pazienti che hanno sviluppato un anticorpo, 25 avevano meno di 20 giorni di esposizione, prima della conferma del risultato. I tempi intermedi fino allo sviluppo di anticorpi con alto titolo o basso titolo erano di 9,0 giorni di esposizione (intervallo 4-24 giorni) o di 12,0 giorni di esposizione (intervallo 6-34 giorni), rispettivamente. Nei pazienti con mutazioni non zero del gene F8 non sono stati riscontrati anticorpi neutralizzanti.
In generale, tutti i pazienti trattati con prodotti a base di fattore VIII della coagulazione devono essere attentamente monitorati per lo sviluppo di anticorpi mediante un'accurata osservazione clinica ed esami di laboratorio. Se non si ottengono i livelli plasmatici di attività del fattore VIII attesi o se l'emorragia non è controllata con una dose adeguata, deve essere eseguito un esame al fine di determinare se siano presenti anticorpi del fattore VIII. Nei pazienti con livelli elevati di anticorpi, è possibile che la terapia con fattore VIII non sia efficace e devono essere prese in considerazione altre soluzioni terapeutiche, come ad esempio l'induzione di immunotolleranza (ITI). Il trattamento di questi pazienti deve essere affidato a medici con esperienza nella gestione dell'emofilia con presenza di anticorpi del fattore VIII.
Complicazioni correlate a catetere
Se è necessario l'uso di un dispositivo di accesso venoso centrale (CVAD), si deve prendere in considerazione il rischio di complicazioni correlate al CVAD, tra cui infezioni locali, batteremia e trombosi in sede di catetere.
Si raccomanda caldamente di annotare il nome e il numero di lotto ogni volta che Nuwiq viene somministrato a un paziente onde disporre di un collegamento fra il paziente e il lotto di medicinale.
Bambini e adolescenti
Le avvertenze e le precauzioni elencate valgono per adulti e bambini in ugual misura.
Considerazioni correlate ad altre sostanze ausiliarie (contenuto di sodio)
Questo medicinale contiene meno di 1 mmol di sodio (23 mg) per flaconcino, ovvero è povero di sodio.
Tuttavia, a seconda del peso corporeo e della posologia, un paziente può ricevere più di un flaconcino (si veda la rubrica «Forma farmaceutica e quantità di principio attivo per unità» per il contenuto di ogni flaconcino). Ciò deve essere considerato dai pazienti che seguono una dieta a basso contenuto di sodio.
Interazioni
Non sono stati effettuati studi d'interazione con Nuwiq.
Gravidanza/Allattamento
Gravidanza
Non sono disponibili dati sufficienti in merito alla gravidanza.
Non vi sono sufficienti studi sulla riproduzione negli animali, sullo sviluppo embrionale, sullo sviluppo del feto e/o sullo sviluppo postnatale. Non si conosce il potenziale rischio per le persone in questo ambito.
Sulla base della rara manifestazione dell'emofilia A nella donna, l'esperienza relativa all'uso di Nuwiq durante la gravidanza non è disponibile. Pertanto, Nuwiq deve essere usato durante la gravidanza solo se chiaramente indicato.
Allattamento
Sulla base della rara manifestazione dell'emofilia A nella donna, l'esperienza relativa all'uso di Nuwiq durante l'allattamento non è disponibile. Pertanto, Nuwiq deve essere usato durante l'allattamento solo se chiaramente indicato
Fertilità
Non sono disponibili dati in merito agli effetti sulla fertilità.
Effetti sulla capacità di condurre veicoli e sull'impiego di macchine
Nuwiq non ha effetti sulla capacità di guidare veicoli o sulla capacità di utilizzare macchine.
Effetti indesiderati
Sintesi del profilo di sicurezza
Sono state osservate raramente reazioni di ipersensibilità o allergiche (che possono comprendere angioedema, bruciore e sensazione urticante in sede di infusione, brividi, vampate, orticaria e orticaria generalizzata, cefalea, ipotensione, letargia, nausea, irrequietezza, tachicardia, costrizione toracica, formicolio, vomito, rantolo), che in alcuni casi possono evolvere in grave anafilassi (incluso shock).
I pazienti affetti da emofilia A possono sviluppare anticorpi neutralizzanti (inibitori) verso il fattore VIII. Un tale sviluppo di anticorpi si manifesta sotto forma di insufficiente risposta clinica. In questi casi, si raccomanda di contattare un centro specializzato per l'emofilia.
Tabella delle reazioni avverse
Durante uno studio clinico con Nuwiq in pazienti pediatrici (età 2-11 anni, n = 58), adolescenti (età 12-17 anni, n = 3) e adulti (n = 129) affetti da emofilia A grave e trattati in precedenza, è stato segnalato un totale di 12 reazioni avverse a farmaci (ADR) (8 negli adulti, 4 nei bambini) manifestatesi in 8 pazienti (4 adulti, 4 bambini). Durante lo studio clinico condotto su 108 pazienti (età 0-146 mesi) affetti da emofilia A grave e non trattati in precedenza, sono state segnalate le seguenti ADR: anticorpi neutralizzanti contro il fattore VIII (inibitori) in 28 pazienti, piressia in 20 pazienti, esantema in 5 pazienti, ipersensibilità in 2 pazienti, e brividi e orticaria rispettivamente in un paziente ciascuno.
La Tabella 1 presentata qui di seguito si basa sulla Classificazione per sistemi e organi secondo MedDRA (SOC e Termini preferiti).
Le frequenze sono state valutate sulla base dei criteri seguenti: molto comune (≥1/10); comune (≥1/100, <1/10); occasionale (≥1/1000, <1/100); raro (≥1/10'000, <1/1000); molto raro (<1/10'000), non nota (la frequenza non può essere definita sulla base dei dati disponibili).
All'interno di ciascuna categoria di frequenza, gli effetti indesiderati sono elencati in ordine di gravità decrescente.
Tabella 1. Frequenza dell'insorgenza di reazioni avverse a farmaci (ADR) per paziente negli studi clinici in 298 pazienti affetti da emofilia A grave e già trattati in precedenza
Classificazione per sistemi e organi secondo MedDRA | Effetti indesiderati | Frequenza |
Anomalie del sistema linfatico e sanguigno | Anticorpi anti fattore VIII | Occasionale (per pazienti trattati in precedenza)# Molto frequente (per pazienti non pretrattati)# |
Patologie del sistema immunitario | Ipersensibilità | Occasionale* |
Patologie del sistema nervoso | Parestesia; Cefalea | Occasionale* |
Patologie dell'orecchio e del labirinto | Vertigine | Occasionale* |
Patologie gastrointestinali | Bocca secca | Occasionale* |
Patologie del sistema muscoloscheletrico e del tessuto connettivo | Dolore dorsale | Occasionale* |
Patologie sistemiche e condizioni relative alla sede di somministrazione | Piressia; Infiammazione in sede di iniezione; Dolore in sede di iniezione Brividi; Malessere | Frequente* Occasionale* |
Esami diagnostici | Positività agli anticorpi non neutralizzanti del fattore VIII (in pazienti precedentemente trattati) | Occasionale* |
Anomalie di vie respiratorie, gabbia toracica e mediastino | Dispnea | Occasionale* |
Patologie di derma e ipoderma | Esantema; Orticaria | Frequente* Occasionale* |
* Tutte queste ADR sono state calcolate su un numero complessivo di 298 pazienti sottoposti agli studi clinici, di cui 190 pazienti precedentemente sottoposti a trattamento e 108 pazienti non sottoposti precedentemente a trattamento.
# La frequenza si basa su studi clinici con tutti i prodotti FVIII che hanno incluso pazienti affetti da emofilia A grave.
Descrizione di reazioni avverse selezionate
È stato segnalato lo sviluppo di anticorpi non neutralizzanti del fattore VIII in un paziente adulto (vedere Tabella 1). Il campione è stato sottoposto ad analisi dal laboratorio centrale in 8 diluizioni. Il risultato è stato positivo solo con il fattore di diluizione 1 e il titolo anticorpale era molto basso. L'attività inibitoria, secondo la misurazione con metodo Bethesda modificato, non è stata rilevata in questo paziente. L'efficacia clinica e il tasso di recupero in vivo di Nuwiq non sono stati inficiati in questo paziente.
Bambini e adolescenti
Si suppone che frequenza, tipo e gravità delle reazioni avverse nei bambini siano identici a quelli degli adulti.
La notifica del sospetto di effetti collaterali, a seguito della registrazione, è di grande importanza. Consente un monitoraggio costante del rapporto rischi/benefici del medicinale. Il personale medico e paramedico è tenuto a indicare ogni sospetto di un nuovo effetto collaterale o di un effetto collaterale grave sul portale online ElViS (Electronic Vigilance System). Informazioni a riguardo sono disponibili su www.swissmedic.ch.
Posologia eccessiva
Non sono stati riportati casi di sovradosaggio.
Proprietà/Effetti
Codice ATC
B02BD02
Meccanismo d'azione
Simoctocog alfa (fattore VIII della coagulazione (rDNA) di natura umana) è una proteina purificata che possiede 1440 amminoacidi. La sequenza degli amminoacidi è simile alla forma da 90 e da 80 kDa del fattore VIII del plasma umano (vale a dire, con delezione del dominio B). Nuwiq è prodotto con la tecnologia del DNA ricombinante nelle cellule 293F di rene embrionale umano (HEK) geneticamente modificate. Al processo di produzione o al medicinale finale non sono aggiunti materiali di derivazione animale o umana.
L'emofilia A è una malattia ereditaria della coagulazione del sangue legata al cromosoma X, con ridotti livelli del fattore VIII:C, che causa emorragie massicce a carico delle articolazioni, dei muscoli o di organi interni, sia spontaneamente che in conseguenza di infortuni o interventi chirurgici. I livelli plasmatici di fattore VIII vengono aumentati per mezzo della terapia sostitutiva, consentendo così la correzione temporanea della carenza di fattore VIII e della tendenza all'emorragia.
Farmacodinamica
Il complesso fattore VIII/Fattore di von Willebrand è formato da due molecole (fattore VIII e fattore von Willebrand) con differenti funzioni fisiologiche. A seguito di infusione in un paziente affetto da emofilia A, il fattore VIII si lega al fattore von Willebrand nella circolazione ematica del paziente. Il fattore VIII attivato agisce come co-fattore per il fattore IX attivato, accelerando la conversione del fattore X in fattore X attivato. Il fattore X attivato converte la protrombina in trombina. La trombina a sua volta converte il fibrinogeno in fibrina dando luogo alla formazione di un coagulo.
Efficacia clinica
L'immunogenicità di Nuwiq è stata valutata in studi clinici in 135 pazienti affetti da emofilia A grave e già trattati in precedenza (74 pazienti adulti e 61 pazienti pediatrici). In 4 pazienti sono stati rilevati anticorpi non neutralizzanti del fattore VIII. 3 pazienti su 4 avevano anticorpi non neutralizzanti già prima della somministrazione di Nuwiq. È stato segnalato lo sviluppo di anticorpi non neutralizzanti del fattore VIII in un paziente adulto. Il campione è stato sottoposto ad analisi dal laboratorio centrale in 8 diluizioni. Il risultato è stato positivo solo con il fattore di diluizione 1 e il titolo anticorpale era molto basso. L'attività inibitoria, secondo la misurazione con metodo Bethesda modificato, non è stata rilevata in questo paziente. L'efficacia clinica e il tasso di recupero in vivo di Nuwiq non sono stati inficiati in questo paziente.
In uno studio clinico condotto su 32 pazienti adulti affetti da emofilia A grave, il consumo mediano di Nuwiq per la profilassi è stato di 468,7 UI/kg/mese. La dose mediana per il trattamento di episodi emorragici importanti è stata di 33,0 UI/kg in questi pazienti trattati in regime di profilassi. In un altro studio clinico, 22 pazienti adulti sono stati trattati al bisogno. In totale sono stati trattati 986 episodi emorragici con una dose mediana di 30,9 UI/kg. In generale, gli episodi emorragici minori hanno richiesto una dose mediana leggermente inferiore, ed episodi più severi hanno richiesto una dose mediana fino a tre volte superiore.
Profilassi personalizzata: la profilassi personalizzata su base PK è stata studiata in 66 pazienti adulti con PTP (piano terapeutico personalizzato) affetti da emofilia A grave. Dopo una fase di profilassi standard della durata di 1-3 mesi (dosaggio ogni due giorni o 3 volte alla settimana), per 44 pazienti (67%) si è potuto cambiare lo schema di dosaggio sulla base del valore PK; 40 pazienti hanno terminato la profilassi della durata di 6 mesi seguendo lo schema di dosaggio calcolato consigliato. Di questi pazienti 34 (85%) sono stati sottoposti a trattamento due volte alla settimana o con frequenza minore. In 33 pazienti (82,5%) non si sono verificate emorragie e 36 (90,0%) non hanno avuto emorragie spontanee. La percentuale annualizzata di emorragie (ABR) (valore medio ± DS) era di 1,2 ± 3,9 e il dosaggio medio ± DS era di 52,2 ± 12,2 UI/kg per iniezione o 99,7 ± 25,6 UI/kg a settimana.
Si deve osservare che l'ABR tra le diverse concentrazioni di fattori e tra i diversi studi clinici non è paragonabile.
Sicurezza ed efficacia nei pazienti pediatrici
I dati seguenti sono stati ottenuti da pazienti pediatrici già trattati in precedenza: 29 bambini di età fra 2 e 5 anni, 31 bambini di età fra 6 e 12 anni e un adolescente di 14 anni. La dose mediana per infusione profilattica è stata di 37,8 UI/kg. Venti pazienti hanno usato dosi mediane di oltre 45 UI/kg. Il consumo mediano di Nuwiq per la profilassi al mese è stato di 521,9 UI/kg. Per trattare gli episodi emorragici nei bambini (43,9 UI/kg) è stata necessaria una dose mediana più alta di Nuwiq che negli adulti (33,0 UI/kg), e per trattare gli episodi emorragici da moderati a maggiori è stata necessaria una dose mediana più alta che per il trattamento degli episodi emorragici minori (78,2 UI/kg vs. 41,7 UI/kg). In generale per bambini piccoli sono state necessarie dosi mediane più alte (6-12 anni: 43,9 UI/kg; 2-5 anni: 52,6 UI/kg). Questi dati sono stati confermati dopo l'osservazione duratura di 49 di questi bambini che sono stati sottoposti a trattamento per un periodo medio supplementare di 30 mesi (intervallo compreso tra 9,5 e 52 mesi); in questo periodo di tempo il 45% dei bambini non ha avuto emorragie spontanee.
Farmacocinetica
Assorbimento
Non applicabile.
Distribuzione
FVIII si distribuisce nel plasma.
Metabolismo
Non applicabile.
Eliminazione
Vedi tabelle 2-4.
Tabella 2. Parametri di farmacocinetica (PK) per Nuwiq (dose: 50 UI/kg) in pazienti adulti trattati in precedenza (età 18-65 anni) affetti da emofilia A grave (n = 20)
Parametro di PK | Metodo cromogenico | Saggio di coagulazione monofase |
|---|---|---|
Media ± DS | Media ± DS | |
AUCnorm (h*UI/ml/(UI/kg)) | 0,39 ± 0,14 | 0,37 ± 0,11 |
T½ (h) | 14,7 ± 10,4 | 17,0 ± 11,8 |
IVR (%/UI/kg) | 2,5 ± 0,4 | 2,2 ± 0,3 |
CL (ml/h/kg) | 3,0 ± 1,2 | 2,9 ± 1,0 |
AUC = area sotto la curva (FVIII:C), T½ = emivita terminale,
IVR = recupero incrementale in vivo, CL = clearance, DS = deviazione standard
Cinetica di gruppi di pazienti speciali
Sottogruppi adeguati in base al peso
Nei pazienti adulti sovrappeso (IMC 25-30 kg/m2) e obesi (IMC >30 kg/m2) inclusi nello studio PK, la clearance è stata superiore rispetto ai pazienti normopeso.
Bambini e adolescenti
Tabella 3. Parametro PK per Nuwiq (dose: 50 UI/kg) in bambini trattati in precedenza (età 6-12 anni) affetti da emofilia A grave (n = 12)
Parametro di PK | Metodo cromogenico | Saggio di coagulazione monofase |
|---|---|---|
Media ± DS | Media ± DS | |
AUCnorm (h*UI/ml/(UI/kg)) | 0,25 ± 0,1 | 0,26 ± 0,1 |
T½(h) | 10,0 ± 1,9 | 13,1 ± 2,6 |
IVR (%/UI/kg) | 1,9 ± 0,4 | 1,6 ± 0,4 |
CL (ml/h/kg) | 4,3 ± 1,2 | 4,1 ± 0,9 |
AUC = area sotto la curva (FVIII:C), T1/2 = emivita terminale,
IVR = recupero incrementale in vivo, CL = clearance, DS = deviazione standard
Tabella 4. Parametri di PK per Nuwiq (dose: 50 UI/kg) in bambini trattati in precedenza (età 2-5 anni) affetti da emofilia A grave (n = 13)
Parametro di PK | Metodo cromogenico | Saggio di coagulazione monofase |
|---|---|---|
Media ± DS | Media ± DS | |
AUCnorm (h*UI/ml/(UI/kg)) | 0,22 ± 0,1 | 0,22 ± 0,1 |
T½(h) | 9,5 ± 3,3 | 11,9 ± 5,4 |
IVR (%/UI/kg) | 1,9 ± 0,3 | 1,6 ± 0,2 |
CL (ml/h/kg) | 5,4 ± 2,4 | 5,4 ± 2,3 |
AUC = area sotto la curva (FVIII:C), T½ = emivita terminale,
IVR = recupero incrementale in vivo, CL = clearance, DS = deviazione standard
Come è noto dalla letteratura medica, il recupero e l'emivita sono stati inferiori nei bambini piccoli rispetto agli adulti mentre la clearance è risultata superiore, fatto che può essere in parte dovuto alla nota maggiore entità del volume plasmatico per chilogrammo di peso corporeo nei pazienti più giovani.
Dati preclinici
Dagli studi sulla tossicità è stato dimostrato che la somministrazione endovenosa locale e l'esposizione sistemica sono state ben tollerate negli animali da laboratorio (ratti e macaco cinomolgo). Non è chiaro il significato del calo di peso del timo osservato nelle scimmie dopo ripetute somministrazioni di simoctocog alfa.
A causa della risposta immunitaria alle proteine eterologhe in tutte le specie di mammiferi non umani non sono stati condotti studi specifici con somministrazione per un periodo prolungato (come ad es. studi su tossicità per la riproduzione, tossicità cronica, cancerogenicità).
Non sono stati condotti studi sul potenziale mutageno di Nuwiq.
Le valutazioni ex vivo, con l'ausilio di un kit commerciale per il test, per quantificare la risposta dei linfociti T alla terapia con proteine hanno dimostrato un'immunogenicità pronunciata almeno quanto quella di preparati paragonabili.
Altre indicazioni
Incompatibilità
Poiché per questo medicamento non sono stati condotti studi di compatibilità, non lo si deve somministrare in combinazione con altri medicamenti.
Si devono usare solo i set per iniezione forniti in dotazione in quanto si può verificare un fallimento terapeutico quale conseguenza dell'assorbimento del fattore VIII della coagulazione di natura umana sulle superfici interne di alcuni dispositivi per iniezione.
Stabilità
Il medicinale non deve essere utilizzato oltre la data indicata con «EXP» sul contenitore.
Durante il periodo di stabilità, il prodotto può essere conservato a temperatura ambiente (fino a 25 °C) per un periodo non superiore a 1 mese. Una volta prelevato dal frigorifero, il medicinale non deve più essere riposto in frigorifero. Annotare l'inizio del periodo di conservazione a temperatura ambiente sulla confezione del prodotto. Conservare il flaconcino nella confezione originale per proteggerlo dalla luce.
Dopo la ricostituzione
Dal punto di vista microbiologico, il prodotto va usato immediatamente dopo la ricostituzione. Se ciò non avviene, il preparato ricostituito deve essere utilizzato entro 3 ore a una temperatura di conservazione di ≤25 °C. Il preparato ricostituito deve essere conservato nel flaconcino.
I preparati inutilizzati che sono stati conservati a temperatura ambiente per più di 3 ore devono essere smaltiti.
Indicazioni particolari concernenti l'immagazzinamento
Conservare in frigorifero (2-8 °C).
Non congelare.
Conservare nella confezione originale per proteggere il contenuto dalla luce.
Tenere fuori dalla portata dei bambini.
Per le condizioni di conservazione del preparato ricostituito, vedere «Altre indicazioni: Stabilità – Dopo la ricostituzione».
Smaltimento
Il medicinale non utilizzato e i rifiuti derivati da tale medicinale devono essere smaltiti in conformità alla normativa locale vigente.
Indicazioni per la manipolazione
La polvere deve essere ricostituita unicamente con il solvente fornito (2,5 ml di acqua per preparazioni iniettabili) usando il set per iniezione fornito. Far oscillare il flaconcino con delicatezza finché la polvere non si è sciolta. Dopo la ricostituzione, la soluzione deve essere riaspirata nella siringa.
Il medicinale ricostituito deve essere controllato visivamente per rilevare eventuali particolati e cambiamenti di colore prima della somministrazione. Il medicinale ricostituito è una soluzione limpida e incolore, priva di particelle estranee e con pH di 6,5-7,5. Non usare soluzioni torbide o con depositi.
Ricostituzione
- Lasciare che la siringa con il solvente (acqua per preparazioni iniettabili) e la polvere nel flaconcino chiuso raggiungano la temperatura ambiente. È possibile farlo tenendoli fra le mani finché non ne avranno raggiunto lo stesso grado di calore. Non riscaldare il flaconcino e la siringa preriempita in nessun altro modo. Questa temperatura va mantenuta durante la ricostituzione.
- Rimuovere la capsula di chiusura a strappo dal flaconcino della polvere esponendo la parte centrale del tappo in gomma. Non rimuovere il tappo grigio o l'anello metallico attorno alla parte superiore del flaconcino.

- Strofinare la parte superiore del flaconcino con un tampone monouso imbevuto di alcol. Lasciare che l'alcol si asciughi.
- Togliere il rivestimento in carta dalla confezione dell'adattatore per flaconcino. Non estrarre l'adattatore dalla confezione.

- Collocare il flaconcino di polvere su una superficie piana e tenerlo fermo. Prendere la confezione dell'adattatore e collocare l'adattatore per flaconcino sul centro del tappo in gomma del flaconcino di polvere. Spingere energicamente la confezione dell'adattatore finché la punta dell'adattatore non penetra nel tappo in gomma. L'adattatore si aggancia al flaconcino quando ciò avviene.

- Togliere il rivestimento in carta dalla confezione della siringa preriempita. Afferrare lo stantuffo all'estremità e non toccare il corpo dello stantuffo. Avvitare l'estremità filettata dello stantuffo al corpo della siringa con il solvente. Girare lo stantuffo in senso orario finché non si incontra una lieve resistenza.

- Togliere la punta di plastica antimanomissione dalla siringa con il solvente staccando la perforazione della capsula di chiusura. Non toccare l'interno della capsula di chiusura o la punta della siringa. Nel caso in cui la soluzione non venisse usata immediatamente, chiudere la siringa riempita con il tappo antimanomissione in plastica per conservarla.

- Togliere la confezione dell'adattatore e gettarla.
- Unire saldamente la siringa con il solvente all'adattatore per flaconcino girando in senso orario finché non si incontra resistenza.

- Iniettare lentamente tutto il solvente nel flaconcino della polvere abbassando lo stantuffo.

- Senza rimuovere la siringa, muovere o girare delicatamente il flaconcino alcune volte con movimento circolare per sciogliere la polvere. Non agitare. Aspettare finché la polvere non si sarà sciolta del tutto.
- Prima di procedere alla somministrazione, controllare visivamente se la soluzione finale contiene particelle. La soluzione deve essere limpida e incolore, priva di particelle visibili. Non usare soluzioni torbide o con depositi.
- Capovolgere il flaconcino fissato alla siringa e aspirare lentamente la soluzione finale nella siringa. Assicurarsi che l'intero contenuto del flaconcino sia trasferito nella siringa.

- Svitare la siringa riempita dall'adattatore per flaconcino girando in senso antiorario e gettare il flaconcino vuoto.
- Ora la soluzione è pronta per essere usata immediatamente. Non conservare in frigorifero.
- Pulire il sito di iniezione scelto con uno dei tamponi imbevuti di alcol forniti in dotazione.
- Fissare alla siringa il set per infusione fornito. Inserire l'ago dell'infusione nella vena scelta. Se è stato usato un laccio emostatico per agevolare l'individuazione della vena, questo va tolto prima di iniziare a iniettare la soluzione. Nella siringa non deve entrare sangue in quanto c'è il rischio che si formino coaguli di fibrina.
- Iniettare la soluzione nella vena con una bassa velocità di infusione, non superiore a 4 ml al minuto.
Se si usa più di un flaconcino di polvere per un trattamento è possibile riutilizzare lo stesso ago. L'adattatore per flaconcino e la siringa sono monouso.
Numero dell'omologazione
65551 (Swissmedic)
Titolare dell’omologazione
Octapharma AG, 8853 Lachen.
Stato dell'informazione
Settembre 2020.
Composition
Principe actif
Simoctocog alfa (facteur VIII de coagulation humain recombinant).
Excipients
Saccharose, chlorure de sodium, chlorure de calcium dihydraté, hydrochlorure d'arginine, citrate de sodium dihydraté, poloxamère 188 (soit une teneur totale en sodium de 18,4 mg/flacon).
Solvant: eau pour préparations injectables.
Forme pharmaceutique et quantité de principe actif par unité
Poudre et solvant pour solution injectable.
Chaque flacon contient nominalement 250/500/1000/2000 UI de facteur VIII de coagulation humain (ADNr), Simoctocog alfa. Une fois reconstitué, Nuwiq contient environ 100/200/400/800 UI par ml de facteur VIII de coagulation humain (ADNr), Simoctocog alfa.
Poudre: poudre friable de couleur blanche à blanc cassé.
Solution eau pour préparations injectables; solution limpide, incolore.
L'activité (UI) a été mesurée en utilisant le test chromogène conformément à la pharmacopée européenne. L'activité spécifique de Nuwiq s'élève à environ 9500 UI/mg de protéine.
Indications/Possibilités d’emploi
Traitement et prophylaxie des hémorragies chez les patients atteints d'hémophilie A (déficit congénital en facteur VIII).
Nuwiq ne contient pas de quantités pharmacologiquement actives de facteur de von Willebrand et il n'est donc pas adapté au traitement du syndrome de von Willebrand-Jürgens.
Posologie/Mode d’emploi
Le traitement par Nuwiq sera initié sous la surveillance d'un médecin expérimenté dans la prise en charge des patients atteints d'hémophilie.
Posologie usuelle
La posologie et la durée du traitement de substitution dépendent de la gravité du déficit en facteur VIII, de la localisation et de l'ampleur de l'hémorragie, ainsi que de l'état clinique du patient.
Le nombre d'unités de facteur VIII à administrer est exprimé en Unités Internationales (U.I.), selon le standard actuel de l'OMS pour les concentrés de facteur VIII. L'activité plasmatique du FVIII est exprimée soit en pourcentage (par rapport au plasma humain normal), soit en Unités Internationales (par rapport au standard international pour le FVIII plasmatique). Le test de coagulation en une seule étape tout comme la méthode chromogénique sont appropriés pour mesurer l'activité du FVIII dans le plasma. Les données du test de coagulation en une seule étape sont généralement utilisées dans l'application clinique et pour la comparaison de l'activité avec d'autres préparations de FVIII recombinant et plasmatique.
Une Unité Internationale (U.I.) du FVIII humain est équivalente à la quantité de FVIII présente dans 1 millilitre de plasma humain.
Traitement à la demande
Le calcul de la dose de facteur VIII nécessaire repose sur le résultat empirique qu'une Unité Internationale (UI) de facteur VIII par kg de poids corporel augmente l'activité plasmatique du facteur VIII en moyenne de 2% par rapport à l'activité normale ou de 2 UI/dl. La dose requise est calculée selon la formule suivante:
- Unités nécessaires = poids corporel (kg) x augmentation souhaitée du facteur VIII (%) (UI/dl) x 0,5 (UI/kg par UI/dl)
- Augmentation attendue du taux de facteur VIII (% de la valeur normale) = 2 x UI administrées : poids corporel (kg)
La dose et la fréquence d'administration doivent toujours être adaptées de façon individuelle en tenant compte de l'efficacité clinique.
Dans les cas d'événements hémorragiques suivants, l'activité du facteur VIII ne doit pas descendre en dessous du taux d'activité plasmatique indiqué (en % de la normale ou en UI/dl) pendant la période correspondante. Le tableau suivant peut servir de guide pour la détermination des posologies lors des épisodes hémorragiques et des interventions chirurgicales:
Intensité de l'hémorragie/Type d'intervention chirurgicale | Taux de facteur VIII nécessaire (%) (UI/dl) | Fréquences des injections (heures)/Durée de traitement (jours) |
|---|---|---|
Hémorragie | ||
Début d'hémarthroses, saignements musculaires ou buccaux | 20–40 | Répéter toutes les 12 à 24 heures pendant au moins 1 jour, jusqu'à la fin de l'épisode hémorragique indiquée par la disparition de la douleur ou l'obtention d'une cicatrisation. |
Hémarthroses et hémorragies musculaires plus étendues, ou hématomes | 30–60 | Répéter l'injection toutes les 12 à 24 heures pendant 3 à 4 jours ou plus jusqu'à disparition de la douleur et de l'invalidité aiguë. |
Hémorragies mettant en jeu le pronostic vital | 60–100 | Répéter l'injection toutes les 8 à 24 h jusqu'à disparition du risque vital. |
Intervention chirurgicale | ||
Interventions mineures, y compris extractions dentaires | 30–60 | Toutes les 24 heures, pendant au moins 1 jour, jusqu'à l'obtention d'une cicatrisation |
Interventions chirurgicales majeures | 80–100 (pré- et postopératoires) | Répéter l'injection toutes les 8 à 24 heures jusqu'à obtention d'une cicatrisation adéquate, puis administrer le traitement pendant au moins 7 jours supplémentaires pour maintenir une activité coagulante du facteur VIII entre 30% et 60% (UI/dl). |
Prophylaxie
Les doses habituelles en prophylaxie à long terme des hémorragies chez les patients atteints d'hémophilie A sévère sont de 20 à 40 UI de facteur VIII par kg de poids corporel à des intervalles de 2 à 3 jours. Dans certains cas, en particulier chez les sujets jeunes, il peut s'avérer nécessaire de raccourcir les intervalles entre les doses ou d'augmenter les doses.
Durant le traitement, il est conseillé d'effectuer un contrôle approprié du taux de facteur VIII afin d'ajuster les doses à administrer et la fréquence des injections. Plus particulièrement pour les interventions chirurgicales majeures, un contrôle rigoureux du traitement substitutif par des mesures de la coagulation (activité plasmatique du facteur VIII) est indispensable. Selon les patients, la réponse au traitement par le facteur FVIII peut varier, entraînant des taux de récupération in vivo et des demi-vies différents.
Afin d’assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Patients âgés
Il n'existe que peu d'expérience chez les patients âgés.
Enfants et adolescents
Pour la prévention prolongée des hémorragies chez les patients âgés de moins de 12 ans, une dose de 30 à 50 UI/kg de FVIII tous les 2 jours ou trois fois par semaine est recommandée.
Patients non préalablement traités
La sécurité et l'efficacité de Nuwiq chez des patients non préalablement traités ont été évaluées dans le cadre d'une étude clinique prospective.
Schéma d'administration
Il est recommandé de ne pas administrer plus de 4 ml par minute.
Mode d’administration
Pour administration intraveineuse.
Instructions de reconstitution du médicament avant utilisation, voir le paragraphe «Remarques particulières, remarques concernant la manipulation».
Contre-indications
Hypersensibilité à l'un des principes actifs ou à l'un des excipients mentionnés.
Mises en garde et précautions
Hypersensibilité
Comme avec tout médicament contenant des protéines et administré par voie intraveineuse, des réactions allergiques de type hypersensibilité peuvent être constatées. Nuwiq contient des traces d'autres cellules hôtes humaines distinctes du facteur VIII. Si des symptômes d'hypersensibilité apparaissent, il faut indiquer aux patients d'interrompre immédiatement l'administration du produit et de contacter leur médecin. Les patients doivent être informés des signes précoces de réaction d'hypersensibilité incluant urticaire, urticaire généralisée, oppression thoracique, respiration sifflante, hypotension et choc anaphylactique.
En cas de choc, le traitement médical standard de l'état de choc devra être mis en place.
Inhibiteurs
La formation d'anticorps neutralisants (inhibiteurs) du facteur VIII est une complication connue du traitement des patients atteints d'hémophilie A. Ces anticorps sont habituellement des immunoglobulines IgG dirigées contre l'activité pro-coagulante du FVIII et sont quantifiés en unités Bethesda (UB) par ml de plasma à l'aide d'un test modifié. Le risque de développement des anticorps est corrélé à l'exposition au facteur antihémophilique VIII, ce risque étant maximal dans les 20 premiers jours d'exposition (JE). Dans de rares cas, les anticorps peuvent se développer après les 100 premiers JE.
Des cas de réapparition d'anticorps (à faible titre) ont été observés après le remplacement d'un facteur VIII par un autre, chez des patients préalablement traités ayant plus de 100 JE et des antécédents de développement d'anticorps. Il est donc recommandé de surveiller attentivement tous les patients afin de détecter l'apparition d'un anticorps suite à un changement de médicament.
L'immunogénicité de Nuwiq a été évaluée chez des patients atteints d'hémophilie A sévère (<1% FVIII:C), non préalablement traités, dans le cadre d'un essai clinique ouvert prospectif. Sur les 105 patients ayant reçu Nuwiq et ayant été soumis à un dépistage des anticorps neutralisants au moins une fois après le début du traitement, 17 patients (16,2%) ont développé des anticorps neutralisants à titre élevé et 11 patients (10,5%) ont développé des anticorps neutralisants à faible titre (dont 5 étaient des anticorps transitoires qui ont été éliminés dans le cadre de la poursuite du traitement prophylactique sans modification de la posologie). Sur les 28 patients ayant développé des anticorps, 25 présentaient moins de 20 JE avant le dépistage. Les délais moyens de développement des anticorps à titre élevé ou à faible titre étaient respectivement de 9,0 JE (fourchette de 4 à 24 JE) et de 12,0 JE (fourchette de 6 à 34 JE). Aucun anticorps neutralisant n'a été détecté chez les patients présentant des mutations non nulles du gène F8.
De manière générale, tous les patients traités par un facteur VIII de coagulation doivent faire l'objet d'une surveillance étroite pour détecter l'apparition d'anticorps par un suivi clinique et à l'aide de tests biologiques appropriés. Si le taux de facteur VIII plasmatique attendu n'est pas atteint ou si l'hémorragie n'est pas contrôlée par une dose adéquate, un test de détection d'anticorps du facteur VIII doit alors être effectué. Chez les patients présentant un titre élevé d'anticorps, le traitement de substitution en facteur VIII peut ne pas être efficace et d'autres options thérapeutiques, telles qu'un protocole d'induction de tolérance immune (ITI), doivent être considérées. Le suivi de tels patients doit être effectué par des médecins expérimentés dans la prise en charge de l'hémophilie et des anticorps du facteur VIII.
Complications liées au cathéter
Si un dispositif d'accès veineux central (DAVC) est nécessaire, le risque de complications liées au DAVC, notamment des infections locales, une bactériémie et une thrombose au site du cathéter, doit être pris en compte.
À chaque administration de Nuwiq à un patient, il est fortement recommandé d'enregistrer le nom et le numéro de lot du produit afin de maintenir un lien entre le patient et le numéro de lot du médicament.
Population pédiatrique
Les mises en garde et mesures de précaution indiquées s'appliquent également aux enfants et aux adultes.
Remarques sur les autres composants (teneur en sodium)
Ce médicament contient moins de 1 mmol de sodium (23 mg) par flacon, c'est-à-dire qu'il est presque exempt de sodium.
Toutefois, selon son poids et la posologie, il se peut qu'un patient reçoive plus d'un flacon (voir la rubrique «Forme pharmaceutique et quantité de principe actif par unité» pour le contenu par flacon). Cela doit être pris en considération par les patients qui suivent un régime contrôlé en sodium.
Interactions
Aucune étude des interactions de Nuwiq avec d'autres médicaments n'a été réalisée.
Grossesse/Allaitement
Grossesse
Il n'existe pas de données suffisantes concernant l'emploi chez la femme enceinte.
Il n'existe pas d'expérimentations animales suffisantes concernant les effets sur la grossesse, le développement embryonnaire, le développement fœtal et/ou le développement post-natal. Le risque potentiel pour l'être humain n'est pas connu.
En raison de la rareté des cas d'hémophilie A chez les femmes, il n'existe aucune donnée sur l'utilisation de Nuwiq chez les femmes enceintes. Par conséquent, Nuwiq ne doit être utilisé pendant la grossesse qu'en cas de nécessité absolue.
Allaitement
En raison de la rareté des cas d'hémophilie A chez les femmes, il n'existe aucune donnée sur l'utilisation de Nuwiq chez les femmes allaitantes. Par conséquent, Nuwiq ne doit être utilisé pendant l'allaitement qu'en cas de nécessité absolue.
Fertilité
Il n'existe pas de données concernant l'effet sur la fertilité.
Effet sur l’aptitude à la conduite et l’utilisation de machines
Nuwiq n'a aucune influence sur l'aptitude à la conduite ou à l'utilisation de machines.
Effets indésirables
Résumé du profil de sécurité
Dans de rares cas, des réactions d'hypersensibilité ou allergiques (par exemple, angio-œdème, brûlure et piqûre au site d'injection, frisson, rougeur au visage et bouffée de chaleur, urticaire généralisé, céphalées, éruption cutanée, hypotension, léthargie, nausée, agitation, tachycardie, oppression thoracique, picotement, vomissement, respiration sifflante) ont été observées avec les concentrés de facteur VIII. Dans des cas isolés, ces réactions/symptômes peuvent évoluer vers une anaphylaxie sévère (y compris un choc).
Les patients atteints d'hémophilie A peuvent développer des anticorps neutralisants (inhibiteurs) du facteur VIII. L'apparition de tels anticorps se caractérise par une réponse clinique insuffisante. Dans de tels cas, il est recommandé de contacter un centre spécialisé en hémophilie.
Tableau des effets indésirables
Lors d'études cliniques avec Nuwiq chez des enfants (2 à 11 ans, n = 58), des adolescents (12 à 17 ans, n = 3) et des adultes (n = 129) préalablement traités et atteints d'hémophilie A sévère, 12 effets indésirables du médicament (8 chez les adultes et 4 chez les enfants) ont été rapportés au total chez 8 patients (4 adultes et 4 enfants). Dans une étude clinique portant sur 108 patients (âgés de 0 à 146 mois) atteints d'hémophilie A sévère, non préalablement traités, il a été fait état des effets indésirables du médicament suivants: anticorps neutralisants anti-facteur VIII (inhibiteurs) chez 28 patients, pyrexie chez 20 patients, éruption cutanée chez 5 patients, hypersensibilité chez 2 patients ainsi que frissons et urticaire (un patient chacun).
Le Tableau 1 ci-dessous est présenté conformément à la classification des systèmes d'organes MedDRA (CSO et termes préconisés).
La fréquence a été définie selon les critères suivants: très fréquent (≥1/10); fréquent (≥1/100 à <1/10); occasionnel (≥1/1'000 à <1/100); rare (≥1/10'000 à <1/1'000); très rare (<1/10'000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Au sein de chaque groupe de fréquence, les effets indésirables sont indiqués par degré de gravité décroissant.
Tableau 1. Fréquence de survenue des effets indésirables, par patient, lors des études cliniques de 298 patients atteints d'hémophilie A sévère.
Classe de systèmes d'organes MedDRA | Effets indésirables | Fréquence* |
Troubles du système sanguin et lymphatique | Anticorps anti-facteur VIII | occasionnel (chez les patients préalablement traités)# très fréquent (chez les patients non préalablement traités)# |
Affections du système immunitaire | Hypersensibilité | occasionnel* |
Affections du système nerveux | Paresthésies; Céphalées | occasionnel* |
Affections de l'oreille et du labyrinthe | Vertiges | occasionnel* |
Affections gastro-intestinales | Sécheresse buccale | occasionnel* |
Affections musculo-squelettiques et systémiques | Lombalgie | occasionnel* |
Troubles généraux et anomalies au site d'administration | Pyrexie Inflammation au site d'administration; Douleurs au site d'administration; Frissons Malaise | fréquent* occasionnel* |
Investigations | Présence d'anticorps non neutralisants dirigés contre le facteur VIII (chez les patients non préalablement traités) | occasionnel* |
Troubles respiratoires, thoraciques et médiastinaux | Dyspnée | occasionnel* |
Affections cutanées et sous-cutanées | Éruption cutanée Urticaire | fréquent* occasionnel* |
* Calculé comme patients ayant des effets indésirables du médicament sur le nombre total de 298 patients étudiés, dont 190 patients préalablement traités et 108 patients non préalablement traités.
# La fréquence est déterminée d'après des études sur tous les produits FVIII, menées auprès de patients atteints d'hémophilie A sévère.
Description de certains effets indésirables
Un anticorps non neutralisant dirigé contre le facteur VIII a été observé chez un patient adulte (voir le Tableau 1). L'échantillon a été testé par le laboratoire central à 8 dilutions. Le résultat n'était positif qu'au facteur de dilution 1 et le titre d'anticorps était très faible. Aucune activité inhibitrice, telle que mesurée par le test de Bethesda modifié, n'a été détectée chez ce patient. L'efficacité clinique et la récupération in vivo de Nuwiq n'ont pas été affectées chez ce patient.
Population pédiatrique
On peut supposer que la fréquence, la nature et la gravité des effets indésirables chez les enfants soient les mêmes que chez les adultes.
La déclaration de suspicion d'effets indésirables après l'autorisation de mise sur le marché est d'une grande importance. Elle permet de réévaluer en permanence le rapport bénéfice/risque du médicament. Les professionnels de la santé sont invités à signaler tout soupçon d'effet indésirable nouveau ou grave via le portail en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur le site www.swissmedic.ch.
Surdosage
Aucun cas de surdosage n'a été rapporté.
Propriétés/Effets
Code ATC
B02BD02
Mécanisme d'action
Le simoctocog alfa (facteur VIII de coagulation humain [ADNr]) est une protéine purifiée constituée de 1440 acides aminés. La séquence d'acides aminés est comparable à la forme 90 kDa et 80 kDa du facteur VIII plasmatique humain (c'est-à-dire sans le domaine B). Nuwiq est produit par la technique de l'ADN recombinant sur cellules rénales embryonnaires humaines (HEK) 293F génétiquement modifiées. Aucun produit d'origine animale ou humaine n'est ajouté au cours du procédé de fabrication ou au médicament final.
L'hémophilie A est un trouble héréditaire de la coagulation sanguine lié au sexe, dû à une diminution du taux de facteur VIII:C et qui se caractérise par des saignements abondants au niveau des articulations, des muscles ou des organes internes, soit spontanément soit suite à un traumatisme accidentel ou chirurgical. Grâce à un traitement de substitution, le taux plasmatique en facteur VIII est augmenté, ce qui permet ainsi une correction temporaire du déficit en facteur VIII et de la tendance au saignement.
Pharmacodynamique
Le complexe facteur VIII/facteur de von Willebrand se compose de deux molécules (facteur VIII et facteur de von Willebrand) aux fonctions physiologiques distinctes. Lorsqu'il est injecté à un patient atteint d'hémophilie A, le facteur VIII se fixe sur le facteur de von Willebrand présent dans la circulation sanguine du patient. Le facteur VIII activé agit comme cofacteur du facteur IX activé, accélérant la conversion du facteur X en facteur X activé. Le facteur X activé convertit la prothrombine en thrombine. La thrombine convertit ensuite le fibrinogène en fibrine, ce qui aboutit à la formation d'un caillot.
Efficacité clinique
L'immunogénicité de Nuwiq a été évaluée dans des études cliniques réalisées chez 135 patients atteints d'hémophilie A sévère préalablement traités (74 adultes et 61 patients pédiatriques). Des anticorps non neutralisants relatifs au facteur VIII ont pu être détectés chez 4 patients. 3 des 4 patients présentaient déjà des anticorps non neutralisants avant l'administration de Nuwiq. Des anticorps non neutralisants dirigés contre le facteur VIII ont été détectés chez un patient adulte. L'échantillon a été testé au travers de 8 dilutions par un laboratoire centralisé. Uniquement avec un facteur de dilution de 1, le résultat du test était positif et le titre d'anticorps était très faible. Une activité inhibitrice, selon le test de Bethesda modifié, n'a pas pu être décelée chez ce patient. L'efficacité clinique et la récupération in vivo de Nuwiq n'ont pas été altérées chez le patient.
Dans une étude clinique réalisée chez 32 patients adultes atteints d'hémophilie A sévère, la dose mensuelle moyenne de Nuwiq lors du traitement prophylactique était de 468,7 UI/kg/mois. La dose moyenne de traitement d'épisodes hémorragiques était de 33,0 UI/kg pour les saignements intermittents chez les patients recevant un traitement prophylactique. Dans une autre étude clinique, 22 patients adultes ont été traités à la demande. Au total, 986 épisodes hémorragiques ont été traités avec une dose moyenne de 30,9 UI/kg. En règle générale, les saignements mineurs nécessitaient des doses légèrement inférieures, et les saignements plus abondants nécessitaient des doses moyennes jusqu'à trois fois plus élevées.
Prophylaxie individualisée: la prophylaxie individualisée sur base de la PK a été évaluée chez 66 patients adultes déjà traités (PTP) et présentant une hémophilie A sévère. Après une phase de prophylaxie standard de 1 à 3 mois (administration un jour sur deux ou 3 fois par semaine), 44 patients (67%) sont passés à un schéma d'administration basé sur leur évaluation PK, et 40 patients ont terminé les 6 mois de prophylaxie selon le schéma d'administration et de traitement qui leur avait été attribué. Parmi ces patients, 34 (85%) ont été traités deux fois par semaine ou moins. 33 patients (82,5%) n'ont présenté aucune hémorragie et 36 patients (90,0%) n'ont eu aucun saignement spontané. Le taux de saignement annualisé (TSA; moyen ± ET) était de 1,2 ± 3,9 et la dose moyenne ± ET était de 52,2 ± 12,2 UI/kg par injection et 99,7 ± 25,6 UI/kg par semaine. Il est à noter que le taux de saignement annualisé (TSA) n'est pas comparable entre les différents concentrés de facteur et entre les différentes études cliniques.
Sécurité et efficacité chez les patients pédiatriques
Les données ont été obtenues chez 29 enfants préalablement traités âgés de 2 à 5 ans, 31 enfants âgés de 6 à 12 ans et un adolescent de 14 ans. La dose moyenne par perfusion prophylactique était de 37,8 UI/kg. Vingt patients ont utilisé des doses moyennes de plus de 45 UI/kg. La dose moyenne mensuelle de Nuwiq pour la prophylaxie était de 521,9 UI/kg. La dose moyenne de Nuwiq nécessaire pour traiter les hémorragies chez les enfants (43,9 UI/kg) était supérieure à la dose moyenne nécessaire chez les adultes (33,0 UI/kg). Une dose moyenne plus élevée a été nécessaire pour traiter les hémorragies modérées à majeures par rapport aux hémorragies mineures (78,2 UI/kg contre 41,7 UI/kg). Les jeunes enfants ont en général eu besoin de doses moyennes plus élevées (6-12 ans: 43,9 UI/kg; 2-5 ans: 52,6 UI/kg). Ces données ont été corroborées par le suivi à long terme de 49 de ces enfants qui ont bénéficié d'une période de traitement supplémentaire médiane d'environ 30 mois (fourchette de 9,5 à 52 mois). Au cours de cette période, 45 % des enfants n'ont présenté aucun saignement spontané.
Pharmacocinétique
Absorption
Non applicable.
Distribution
Le FVIII se distribue dans le plasma.
Métabolisme
Non applicable.
Élimination
Voir les tableaux 2 à 4.
Tableau 2. Paramètres pharmacocinétiques de Nuwiq (dose: 50 UI/kg) chez des patients adultes préalablement traités (âgés de 18 à 65 ans) atteints d'hémophilie A sévère (n = 20)
Paramètre pharmacocinétique | Méthode chromogénique | Test de coagulation en une étape |
|---|---|---|
Moyenne ± ET | Moyenne ± ET | |
ASC (h*UI/ml/(UI/kg)) | 0,39 ± 0,14 | 0,37 ± 0,11 |
T½ (h) | 14,7 ± 10,4 | 17,0 ± 11,8 |
IVR (%/UI/kg) | 2,5 ± 0,4 | 2,2 ± 0,3 |
CL (ml/h/kg) | 3,0 ± 1,2 | 2,9 ± 1,0 |
ASC = Aire sous la courbe (FVIII:C), T½ = demi-vie terminale,
IVR = récupération in vivo incrémentale, Cl = Clairance, ET = écart-type
Cinétique des groupes de patients particuliers
Sous-groupes ajustés sur le poids
Chez les patients adultes pré-obèses (IMC 25-30 kg/m2) et chez les patients obèses (IMC >30 kg/m2) de l'étude de pharmacocinétique, la clairance était supérieure à celle des patients ayant un poids normal.
Population pédiatrique
Tableau 3. Paramètres pharmacocinétiques de Nuwiq (dose: 50 UI/kg) chez des enfants préalablement traités âgés de 6 à 12 ans atteints d'hémophilie A sévère (n = 12)
Paramètre pharmacocinétique | Méthode chromogénique | Test de coagulation en une étape |
|---|---|---|
Moyenne ± ET | Moyenne ± ET | |
ASC (h*UI/ml/(UI/kg)) | 0,25 ± 0,1 | 0,26 ± 0,1 |
T½(h) | 10,0 ± 1,9 | 13,1 ± 2,6 |
IVR (%/UI/kg) | 1,9 ± 0,4 | 1,6 ± 0,4 |
CL (ml/h/kg) | 4,3 ± 1,2 | 4,1 ± 0,9 |
ASC = Aire sous la courbe (FVIII:C), T½ = demi-vie terminale,
IVR = récupération in vivo incrémentale, Cl = Clairance, ET = écart-type
Tableau 4. Paramètres pharmacocinétiques de Nuwiq (dose: 50 UI/kg) chez des enfants préalablement traités âgés de 2 à 5 ans atteints d'hémophilie A sévère (n = 13)
Paramètre pharmacocinétique | Méthode chromogénique | Test de coagulation en une étape |
|---|---|---|
Moyenne ± ET | Moyenne ± ET | |
ASC (h*UI/ml/(UI/kg)) | 0,22 ± 0,1 | 0,22 ± 0,1 |
T½(h) | 9,5 ± 3,3 | 11,9 ± 5,4 |
IVR (%/UI/kg) | 1,9 ± 0,3 | 1,6 ± 0,2 |
CL (ml/h/kg) | 5,4 ± 2,4 | 5,4 ±2,3 |
ASC = Aire sous la courbe (FVIII:C), T½ = demi-vie terminale,
IVR = récupération in vivo incrémentale, Cl = Clairance, ET = écart-type
Comme la littérature le mentionne, la récupération et la demi-vie étaient plus faibles chez les jeunes enfants que chez les adultes, et la clairance était plus élevée, ce qui peut être dû en partie au fait que le volume plasmatique par kilogramme de poids corporel est supérieur chez les patients plus jeunes.
Données précliniques
Les études toxicologiques ont montré que l'administration intraveineuse locale et l'exposition systémique étaient bien tolérées chez les animaux de laboratoire (rats et singes cynomolgus). L'implication de l'administration répétée chez le singe du Simoctocog alfa dans la diminution de poids du thymus reste incertaine.
En raison de la réponse immunitaire aux protéines hétérologues attendues chez toutes les espèces de mammifères non humaines, il n'a été mené aucune étude spécifique à doses répétées avec Nuwiq sur une période prolongée (telles que des études sur la toxicité de reproduction, la toxicité chronique et la cancérogénicité). Aucune étude sur le potentiel mutagène de Nuwiq n'a été réalisée.
Des études ex vivo effectuées à l'aide d'un kit de test commercial pour quantifier la réponse des lymphocytes T aux protéines thérapeutiques indiquent une immunogénicité au moins aussi marquée que celle des produits comparables.
Remarques particulières
Incompatibilités
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé à d'autres médicaments.
Utiliser exclusivement le dispositif de perfusion fourni, car un échec du traitement peut survenir suite à l'adsorption du facteur VIII de coagulation humain sur les surfaces internes de certains dispositifs de perfusion.
Stabilité
Le médicament ne doit pas être utilisé après la date de péremption indiquée sur l'emballage après la mention «Verw. bis/EXP».
Pendant la durée de conservation, la préparation peut être entreposée à température ambiante (jusqu'à 25 °C) pendant une durée ne dépassant pas 1 mois. Le produit ne doit pas être replacé au réfrigérateur après en avoir été sorti. Veuillez noter la date de début de conservation à température ambiante sur l'emballage. Le flacon doit être conservé dans son emballage d'origine afin de le protéger de la lumière.
Après la reconstitution
Du point de vue microbiologique, le médicament doit être utilisé immédiatement après la reconstitution. À défaut, la solution reconstituée conservée à une température ambiante ≤25 °C doit être utilisée dans un délai de 3 heures. La solution reconstituée doit être conservée dans le flacon.
Les préparations inutilisées, qui sont restées à température ambiante plus de 3 heures, doivent être éliminées.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C).
Ne pas congeler.
Conserver le récipient dans son carton pour le protéger de la lumière.
Tenir hors de portée des enfants.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique «Remarques particulières: conservation — après la reconstitution».
Traitement des déchets
Tout produit non utilisé ou déchet doit être éliminé conformément aux exigences locales.
Remarques concernant la manipulation
La poudre ne doit être reconstituée qu'avec le solvant fourni (2,5 ml d'eau pour préparations injectables) en utilisant le dispositif de perfusion fourni. Le flacon doit être agité doucement par rotation jusqu'à dissolution complète de la poudre. Après reconstitution, la solution est aspirée dans la seringue.
Avant toute administration, inspecter minutieusement le médicament reconstitué pour déceler des particules en suspension ou une décoloration. Le médicament reconstitué est une solution limpide et incolore, exempte de particules étrangères, dont le pH est compris entre 6,5 et 7,5. Ne pas utiliser de solutions troubles ou présentant des dépôts.
Reconstitution
- Amenez à température ambiante la seringue de solvant (eau pour préparations injectables) et la poudre du flacon fermé avant toute utilisation. Pour cela, tenez-les dans vos mains jusqu'à ce qu'elles soient aussi chaudes que vos mains. Ne pas utiliser d'autres moyens pour chauffer le flacon et la seringue. Cette température doit être maintenue au cours de la reconstitution.
- Enlevez l'opercule amovible en plastique du flacon de poudre afin de découvrir la partie centrale du bouchon en caoutchouc. Ne pas retirer le bouchon gris ni la bague métallique (capsule à sertir) entourant la partie supérieure du flacon.

- Nettoyez le dessus du flacon à l'aide d'un tampon imbibé d'alcool. Laissez sécher.
- Retirez le papier protecteur de l'emballage du dispositif de transfert pour flacon. Ne pas retirer le dispositif de son emballage.

- Placez et maintenez le flacon de poudre sur une surface plane. Tout en tenant l'emballage du dispositif de transfert, placez le dispositif au-dessus du centre du bouchon en caoutchouc du flacon de poudre. Appuyez fermement sur l'emballage du dispositif de transfert jusqu'à ce que la pointe du dispositif pénètre dans le bouchon en caoutchouc. Une fois l'opération terminée, le dispositif de transfert s'enclenche sur le flacon.

- Retirez le papier protecteur de l'emballage de la seringue préremplie. Tenez la tige du piston par l'extrémité et ne touchez pas l'axe. Fixez l'extrémité filetée de la tige du piston à la seringue de solvant. Faites tourner la tige du piston dans le sens des aiguilles d'une montre jusqu'à ce qu'une légère résistance soit ressentie.

- Retirez l'embout protecteur d'inviolabilité en plastique de la seringue de solvant après avoir rompu la zone perforée du capuchon. Ne pas toucher l'intérieur de l'opercule ni l'embout de la seringue. Si la solution n'est pas utilisée immédiatement, fermez la seringue remplie, au moyen de l'embout, pour la conserver.

- Retirez l'emballage du dispositif de transfert et jetez-le.
- Fixez la seringue de solvant au dispositif de transfert en poussant fermement et en tournant dans le sens des aiguilles d'une montre jusqu'à ce qu'une légère résistance soit ressentie.

- Injectez lentement tout le solvant dans le flacon de poudre en appuyant sur la tige du piston.

- Pour dissoudre la poudre, secouez ou remuez délicatement le flacon circulairement pendant quelques secondes sans extraire la seringue. Ne pas agiter. Attendez que la poudre soit totalement dissoute.
- Avant toute administration, vérifiez visuellement l'absence de particules dans la solution obtenue. La solution doit être limpide, incolore et pratiquement exempte de particules visibles. Ne pas utiliser de solutions troubles ou présentant des dépôts.
- Retournez le flacon fixé à la seringue, et aspirez lentement la solution dans la seringue. Veillez à ce que la totalité du flacon soit transférée dans la seringue.

- Détachez la seringue remplie du dispositif de transfert en tournant dans le sens contraire des aiguilles d'une montre, et jetez le flacon vide.
- La solution est à présent prête pour une utilisation immédiate. Ne pas la mettre au réfrigérateur.
- Nettoyez le site d'injection choisi avec l'un des tampons imbibés d'alcool fournis.
- Fixez la tubulure du nécessaire à perfusion fourni à la seringue. Introduisez l'aiguille du nécessaire à perfusion dans la veine choisie. Si vous avez utilisé un garrot pour rendre la veine plus visible, enlevez le garrot avant de commencer l'injection de la solution. Du sang ne doit pas pénétrer dans la seringue, en raison du risque de formation de caillots de fibrine.
- Injectez lentement la solution dans la veine, le débit ne doit pas excéder 4 ml par minute.
Si vous utilisez plus d'un flacon de poudre au cours d'un même traitement, vous pouvez réutiliser la même aiguille d'injection. Le dispositif de transfert et la seringue sont à usage unique.
Numéro d’autorisation
65551 (Swissmedic).
Titulaire de l’autorisation
Octapharma AG, 8853 Lachen.
Mise à jour de l’information
Septembre 2020.
Отзывов (0)
Вы смотрели


Преднизон Спириг 20 мг 20 таблеток
Что такое Преднизон Спириг и когда он используется?Преднизон Спириг содержит активный ингредиент пре..
2012.38 RUB



Levetiracetam Sandoz Lösung 100mg/ml Flasche 300ml
Was ist Levetiracetam Sandoz und wann wird es angewendet?Levetiracetam Sandoz enthält als Wirkstoff ..
15046.89 RUB



Бесплатная консультация опытного специалиста
Опишите симптомы или нужный препарат – мы поможем подобрать его дозировку или аналог, оформим заказ с доставкой на дом или просто проконсультируем.
Нас 14 специалистов и 0 ботов. Мы всегда будем с вами на связи и сможем связаться в любое время.